
撰文丨李杰,勤浩医药首席科学官
编辑丨于靖
蛋白酶,又被称为“生化剪刀”,通过蛋白质水解将大肽的酰胺键裂解,生成小肽或氨基酸。
蛋白质水解也是许多蛋白质翻译后加工的最后阶段之一,而且是单向进行。蛋白质水解可以在生物合成后立即发生,也可以在蛋白质到达需要发挥作用的部位之后再发生,最终几乎所有的蛋白质都会被水解成氨基酸再利用。人类大约有600种蛋白酶。
几乎所有的生物,甚至一些病毒,都需要蛋白质水解酶才能发挥其作用。蛋白酶形成了一个最大和最多样化的酶家族,曾经它被认为主要都是“消化酶”,现在我们清楚了,蛋白酶参与了生物体功能的各个方面。
“家族成员”包括丝氨酸、半胱氨酸、苏氨酸、谷氨酸和天冬氨酸蛋白酶,它们的作用是促进细胞的蛋白质水解。今天,由于治疗新冠病毒的3CL蛋白酶抑制剂受到极大关注,尤其是辉瑞的nirmatrelvir和盐野义制药的ensitrelvir。在关于蛋白酶抑制剂的系列文章中,我们将回顾其最重要的五类:
(1)治疗高血压的ACE抑制剂
(2)治疗糖尿病的DPP-4抑制剂
(3)治疗艾滋病的HIV蛋白酶抑制剂
(4)治疗HCV的HCV NS3/4A丝氨酸蛋白酶抑制剂
(5)治疗新冠肺炎的3CL蛋白酶抑制剂
本篇文章将回顾血管紧张素转换酶(ACE)抑制剂。
01 肾素–血管紧张素系统(RAS)
肾素,也被称为血管紧张素原酶,是一种介导细胞外体积和动脉血管收缩的酶。[1] 肾素最早于1898年由芬兰生理学家Robert Tigerstedt提出,用来表示一种可溶性因子,在兔体内注射肾提取物后,可以观察到该可溶性因子导致血压急剧上升。
肾素–血管紧张素系统(RAS)是最强大的血压和电解质液体/稳态调节剂之一。它作为血压和心血管功能的主要调节因子被发现,至少为血压的药物干预提供了三个重要的靶点(肾素、ACE和AT1)。[2]
如图1所示,当血压降低时(例如,出血),肾脏释放肾素(一种天冬氨酸蛋白酶)进入循环,催化血管紧张素原转化为血管紧张素Ⅰ。血管紧张素原是一种前酶,是由肝脏产生的α2-球蛋白(452个氨基酸),而血管紧张素Ⅰ是肾脏肾小球旁细胞产生的10肽(10个氨基酸)。
虽然血管紧张素Ⅰ没有升压活性,但它是血管紧张素转换酶(ACE)的底物,ACE通过切断游离C末端的两个氨基酸片段(His-Leu)将血管紧张素Ⅰ转化为血管紧张素Ⅱ。
ACE含有两个同源活性位点,锌离子催化各种蛋白质的C端二肽的裂解,包括血管紧张素Ⅰ和缓激肽。血管紧张素Ⅱ是一种八肽(8个氨基酸),具有生物活性,是人体中最有效的血压升高物。
它与两个膜结合的血管紧张素Ⅱ受体AT1和AT2结合,分别位于肾脏、心脏、血管平滑肌细胞、大脑、肾上腺、血小板、脂肪细胞和胎盘。通过刺激肾上腺皮质上的血管紧张素受体,分泌醛固酮,导致钠潴留,血管收缩,继而使血压升高。这就是为什么RAS有时也被称为肾素–血管紧张素醛固酮系统(RAAS)。
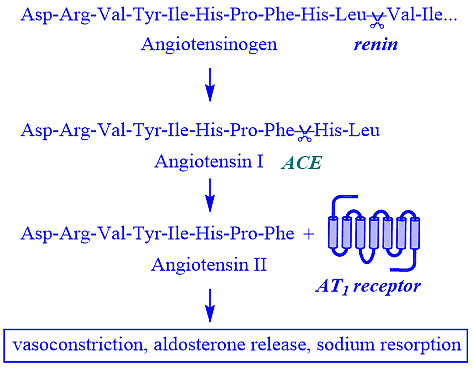
图1:肾素-血管紧张素系统被抑制剂或拮抗剂阻断的部位
02 第一个ACE抑制剂
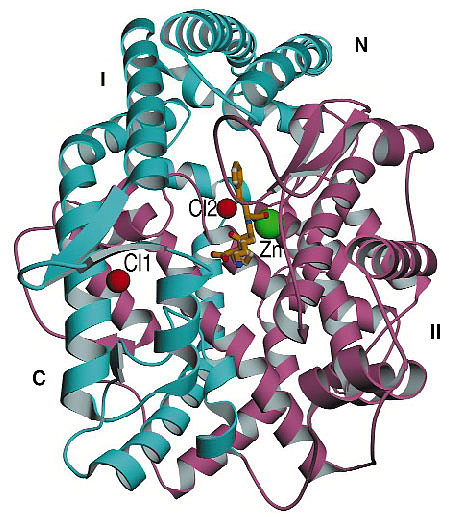
图2:血管紧张素转换酶(ACE)
血管紧张素转换酶(ACE)是一种广泛存在于哺乳动物体内的糖蛋白。尽管ACE有膜结合和游离态两种存在形式,但它主要是血管内皮细胞的一种膜结合酶。ACE是一种蛋白质水解酶。更具体地说,它是一种具有两个同源活性位点的氯依赖性锌金属肽酶,其中锌离子催化各种蛋白质的C端二肽的裂解,包括血管紧张素Ⅰ和缓激肽。[3]
由于血管舒张和盐排泄是缓激肽介导的众多过程之一,ACE对循环缓激肽的破坏也被认为是导致高血压的原因之一。[4]抑制ACE导致的缓激肽积累可能具有积极的血流动力学效应,抑制ACE也会减缓缓激肽的水解,从而刺激前列腺素的合成,但由于该过程缺乏选择性,最终会产生副作用如“干”咳。抑制ACE也可能发生血管性水肿,但较为罕见。[5]
1967年,牛津大学的John Vane将巴西毒蛇Bothrops jararaca毒液的干燥提取物在体外制备成血管紧张素转换酶进行测试,发现它是一种有效的ACE抑制剂。[6]

之后,Vane建议施贵宝研究所将蛇毒提取物作为一种潜在的高血压药物进行研究。在20世纪70年代早期,施贵宝研究所分离出一种泰普罗肽[1,也被称为SQ 20,881,一种九肽(9个氨基酸,见图3)]。在健康志愿者中进行试验,被证明是一种人类选择性ACE抑制剂,可以降低血压。[7]
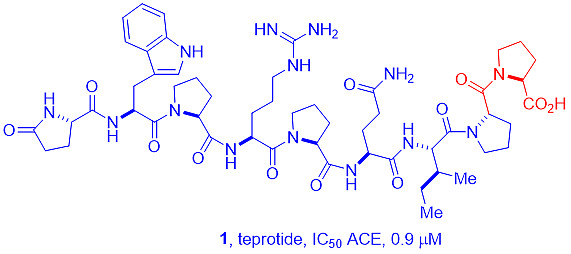
图3:泰普罗肽(1)的结构
David Cushman和Miguel A. Ondetti具有出色的洞察力,他们在施贵宝通过截断泰普罗肽分子,获得琥珀酰-1-脯氨酸(2)分子,ACE抑制IC50为330 nM(见图4)。Cushman和Ondetti选择2作为他们的起点,因为C-末端氨基酸出现在所有天然存在的肽抑制剂的游离C-端。2的活性表明,由于肽键水解的化学性质通常依赖于少量关键氨基酸,小分子量药物只要占据一小部分扩展的活性位点空腔,就可以成为ACE的有效抑制剂。随后,从琥珀酰-1-脯氨酸(2)观察到两个重要发现。
(a)当羧酸被巯基取代时(如硫醇3中的巯基),其活性得到了适度的提高(1.6倍)。
(b)当在酰胺键α-位置安装一个额外的甲基时,结合活性显著提高,生成D-(R)-2-甲基琥珀酰-1-脯氨酸(4),IC50为22 nM。有趣的是,4的L-(S)-异构体活性要低得多。
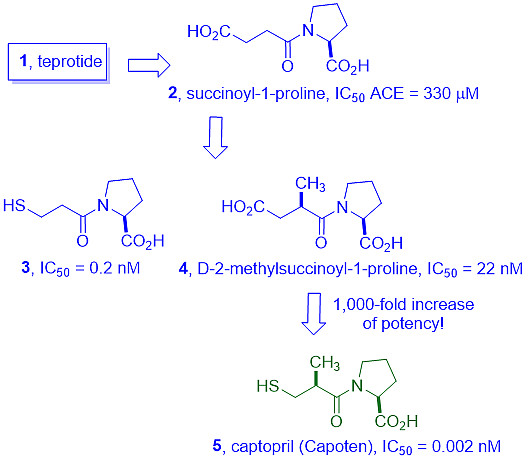
图4:Teprotide(1)进化为卡托普利(5)
当3和4的特征合并到一个分子中时,他们取得了重大突破。因此,他们用巯基(–SH)取代羧酸基,并安装了一个额外的甲基,从而使ACE抑制的效力提高了1000倍。该药成为第一个口服ACE抑制剂卡托普利(captopril,5,Capoten),于1978年获FDA批准,对高血压及高血压相关靶器官损伤的治疗做出重要贡献。施贵宝通过逻辑合成和测试只合成了60个化合物, 并从中找到了卡托普利(5)。[8]
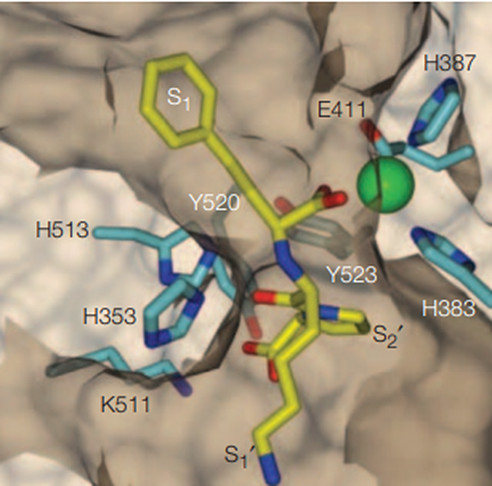
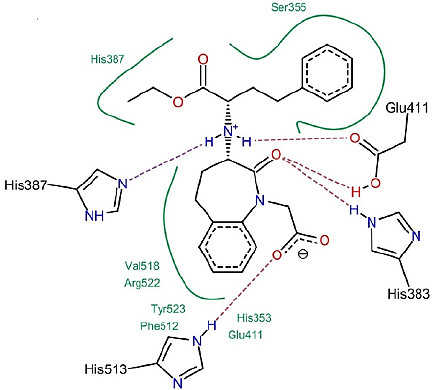
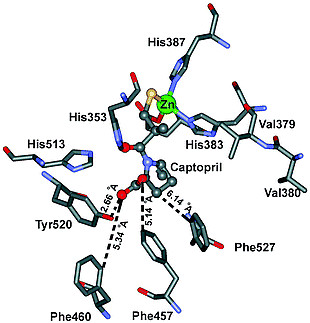
卡托普利(5)成功的意义远远超出了为抑制ACE治疗高血压提供理论依据(CIR)。如图5所示,卡托普利(5)通过螯合临界锌原子发挥作用,从而破坏了酶活性位点的催化成分,而没有填充整个活性位点裂孔。除与活性锌原子形成配位键外,还与酶活性位点内的其他基团形成良好的相互作用关系。进一步的研究表明,卡托普利在ACE酶的S1′和S2′口袋中结合(结合位点的识别见图6)。[9] 这种化合物在抑制血管紧张素Ⅰ的收缩和升压反应方面比泰普罗肽(1)更有效10–20倍。
从动力学上看,卡托普利(5)被认为是一种竞争性酶抑制剂,因为它竞争性地抑制了催化血管紧张素Ⅰ转化为血管紧张素Ⅱ的ACE。随着内源性高血压药物血管紧张素Ⅱ水平的降低,血压下降。
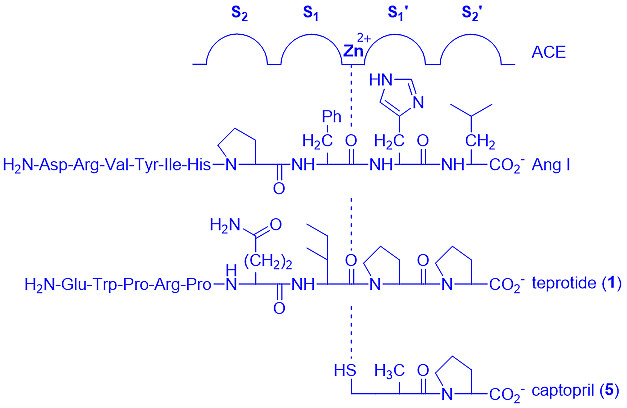
图5:血管紧张素Ⅰ和早期血管紧张素转换酶抑制剂的结合
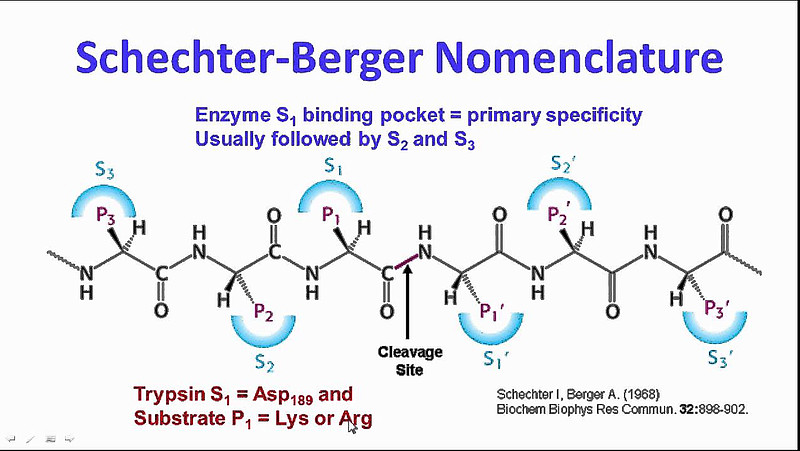
图6:结合位点的识别
卡托普利(5)不仅为十几种“me-too”的ACE抑制剂打开先河,其科学方法也为药物发现开辟了新的途径。它将药物化学从一个对新药的大量经验探索转变为基于机理和设计的真正的科学学科。它是首批应用基于结构的药物设计(SBDD)项目之一,这种方法现在是药物发现的黄金标准。SBDD又称合理药物设计(rational drug design),是一种利用靶点的结构信息,特别是靶点的X-射线结构来改进先导化合物的优化过程,从而加速了药物发现过程的技术。
然而,由于巯基的存在,卡托普利(5)有三个副作用:循环白细胞减少抑制骨髓生长;出现皮疹,丧失味觉;半衰期很短,每天需要服用多次。
03 “Me-too”但“me-better”的ACE抑制剂
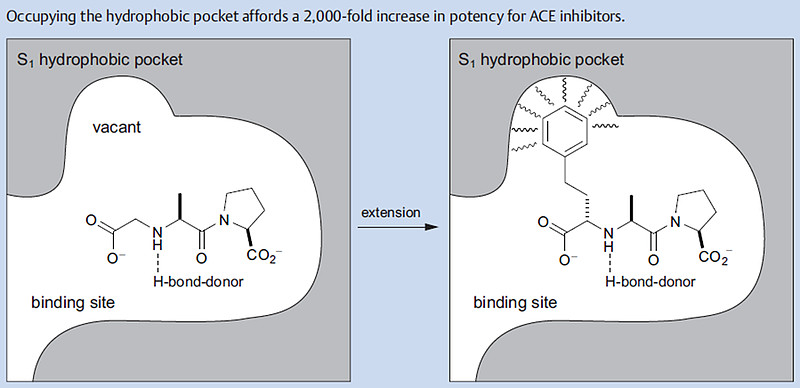
图7:疏水性S1口袋提供了2000倍的效能增长
默沙东召集了一组科学家,由Arthur A. Patchett领导,试图通过改进卡托普利(5)来制造一种更好的ACE抑制剂。他们仔细观察发现,卡托普利(5)并没有完全占据S1口袋(图7)。他们决定用羧酸基团取代这个麻烦的巯基。
围绕通式R-CHCO2H-A1-A2的一系列N-烃羧基二肽进行了广泛的SAR研究。如图8所示,在化合物6中以A1–A2作为Ala–Pro的最优基团进行初步优化。当苄基亚甲基被发现是占据S1袋的最佳组时,突破出现了——ACE抑制的效力增加了2000倍。[10]
结果得到了依那普利拉(7),然而它的口服生物利用度很差。他们只是简单地将酸转化为相应的乙酯,得到依那普利(8,一种依那普利拉的前药)。依那普利(8)具有良好的口服生物利用度,比卡托普利吸收更好。[11]
前药8的另一个优点是延迟作用,这对治疗血压的药物来说是有益的。作用时间变长,每日一次的剂量也足够。由于没有巯基,它也没有与巯基相关的副作用。1985年,默克成功地完成了临床试验,获得批准,并以Vasotec的品牌名称售卖依那普利(8),这也是默克1988年的第一个十亿美元的药物。之后,默克也开发了类似的ACE抑制剂赖诺普利(9,Prinivil,1987年推出),其通式R-CHCO2H–A1–A2中的A1–A2基团是Lys–Pro。
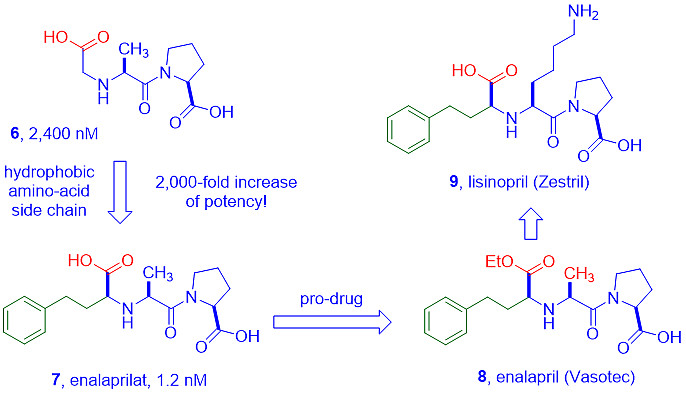
图8:依那普利(8)和赖诺普利(9)的发现路径
04 更多的“me-too”ACE抑制剂
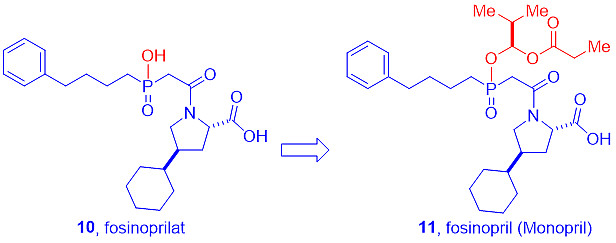
百时美施贵宝(BMS)发现亚磷酸也是一种强效的锌结合剂。这一发现导致福辛普利拉(10)的发现,也是一种强效的 ACE 抑制剂。像依那普利拉(7)一样,福辛普利拉(10)由于其高亲水性,口服生物利用度较差。类似于前药依那普利(8),相对应的前药亚磷酸福辛普利(11,Monopril,1991年推出)具有更高的亲脂性和更好的口服生物利用度。[12]
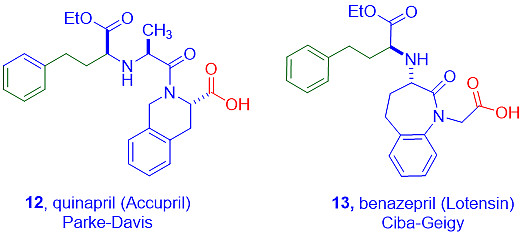
帕克戴维公司(Parke-Davis)后来发现,ACE抑制剂中的脯氨酸环可以扩大到六元环,而不损失任何活性。随后在1991年推出了喹那普利(12, Accupril)。[13] 同年,诺华也推出了他们自己的ACE抑制剂贝那普利(13,Lotensin)。[14]
目前,全世界的药物化学家总共发现了17种ACE抑制剂,它们已经被批准用于临床。这些化合物之间的差异主要是由于结合的活性位点锌和S2′袋的不同。[15]
由于缺乏对ACE抑制的选择性,因此最常见的副作用是“干”咳。[16] 如咳嗽和血管性水肿等副作用,在罕见的情况下,也是ACE抑制减缓缓激肽水解的结果,因为缓激肽水解会刺激前列腺素的合成。
05 经验教训
ACE抑制剂的发现预示着基于结构的药物设计(SBDD)的黄金时代的到来。
从蛇毒中发现卡托普利的过程给我们上了宝贵的一课,即通过截短和去肽化将天然底物转化为药物。虽然所有的ACE抑制剂都有一个与催化活性锌离子螯合的官能团,但默克发现的疏水S1袋提供了BIC的ACE抑制剂。以此为支撑也导致了更多的仿制药出现。即使在今天,ACE抑制剂作为一种治疗高血压的药物,每年的收益仍超过60亿美元。
参考文献:
1、Tigerstedt, R.; Bergman, P. Skand. Arch. Physiol. 1898, 8, 223–271.
2、Opie, L. H. The discovery of captopril: From large animals to small molecules. Cardiovasc. Res. 1995, 30, 18–25
3、Patchett, A. A.; Harris, E.; Tristram, M. J.; Wyvratt, M. J.; Wu, M. T.; Taub, D.; Peterson, E. R.; Ikeler, T. J.; ten Broeke, J.; Payne, L. G.; et al. Nature 1980, 288, 280–283.
4、Ram, C. V. S. Am. J. Cardiovasc. Drugs 2002, 2, 77–89..
5、Skeggs, L. T.; Marsh, W. H.; Khan, J. R.; Shumway, N. P. J. Exp. Med. 1956, 103, 295–299.
6、Smith, C. G.; Vane, J. R. FASEB 2003, 17, 788–789.
7、Cushman, D. W.; Ondetti, M. A. Nature Med. 1999, 5, 1110–1112.
8、Ondetti, M. A.; Williams, N. J.; Sabo, E. F.; Pluscec, J.; Weaver, E. R.; Kocy, O. Biochemistry 1971, 10, 4033–4039.
9、Ondetti, M. A.; Rubin, B.; Cushman, D. Science 1977, 196, 441–444.
10、Patchett, A. A. Enalapril and Lisinopril, In Chronicles of Drug Discovery, Lednicer, D., ed. 1993, pp125–162.
11、Ulm, E. H. Drug Metab. Rev. 1983, 14, 99–110.
12、Krapcho, J.; Turk, C.; Cushman, D. W.; et al. J. Med. Chem. 1988, 31, 1148–1160.
13、Klutchko, S.; Blankley, C. J.; Fleming, R. W.; Hinkley, J. M.; Werner, A. E.; Nording, I.; Holmes, A.; Hoefle, M. L.; Cohen, D. M.; Essenburg, A. D.; Kaplan, H. R. J. Med. Chem. 1986, 29, 1953–1961.
14、Watthey, J. W. H.; Stanton, J. L.; Desai, M.; Babiarz, J. E.; Finn, B. M. J. Med. Chem. 1985, 28, 1511–1516.
15、Zaman, M. A.; Oparil, S.; Calhoun, D. A. Nat. Rev. Drug Discovery 2002, 1, 621–636.
16、Visser, L. E.; Stricker, B. H.; van der Velden, J.; et al. J. Clin. Epidemiol. 1995, 48, 851–857.