书接上回,NASH流水账继续。回顾第一篇请见非酒精性脂肪肝病(NAFLD)流水账之一:疾病概况
2. 药物开发思路
2.1. 主要靶点
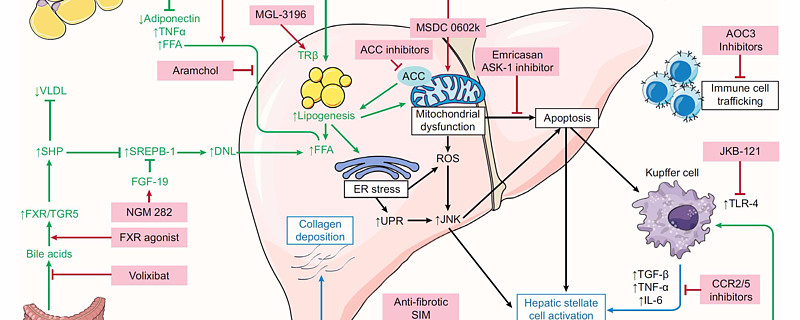
目前药企临床管线的靶点选择思路仍然大体遵循“二次打击”假说,以代谢、炎症和纤维化三个大方向入手,部分靶点对多个方面皆有作用。以现阶段各个靶点管线的临床推进情况来看,作用于下游通路的单一靶点药物、特别是单纯抗纤维化药物,似乎有“治标不治本”之嫌,而作用于更上游通路和多靶点联动的策略可能更有成药潜力。
2.1.1. 代谢类靶点
代谢类靶点主要包括作用于脂代谢的PPAR α/γ/δ激动剂、TRβ受体激动剂、ACC抑制剂、ANGPTL-3抑制剂、FGF-21类似物等,作用于胆酸的FXR抑制剂、FGF-19类似物等,作用于葡萄糖代谢的GLP-1、果糖酶抑制剂、SGLT-2抑制剂、MTOR增敏剂等。
2.1.2. 抗炎症靶点
抗炎症靶点主要以缓解氧化压力、阻断炎症因子和作用于免疫系统等,包括ASK-1、TLR-4、CCR2/5、Caspase、AOC-3抑制剂、NKT细胞抑制剂等。
2.1.3. 抗纤维化靶点
抗纤维化靶点相对较少,包括CCR2/5、ASK-1,Caspase、5-LO等。

[数据来源:Konerman MA et al. J Heptol. 2017; 68(2): 362-75]
2.2. 临床前动物模型
NAFLD作为致病机理都不甚明确的疾病,在药物开发过程中碰上最头疼的问题之一就是难以找到合适的动物模型。目前基本无法找到天然存在的老鼠NASH模型(有一家Taconic Biosciences公司号称可以提供现成的NASH动物模型),只能通过各种手段去一定程度上模拟NASH的症状,而由于背后机理理解的浅薄,很可能出现张冠李戴、模拟出动物体内的机制与人体内真实情况相距甚远的情况,例如有很多模型在组织病理上的变化与临床NASH很相似,但是肝脏的代谢、转录特点可能并不一致。因此动物模型不应仅在组织学上,还应该在蛋白质组、脂质组和转录组等基础上评估模型与临床疾病的相关性,其中一些转录组特征包括免疫信号、脂质代谢、糖代谢改变等以目前水平还比较难模拟。
除了模型种类带来的问题外,目前几乎所有老鼠模型在国内均无法进行肝穿来确定基线疾病状态,即在给药前研究者完全无法确定老鼠究竟处于何种疾病病程,这对临床前药效评价带来很大困扰。
2.2.1. 现有主流老鼠NASH模型
首先是利用与临床肥胖相似的营养结构饮食引起的肥胖模型,在高脂肪饮食诱导的基础上添加高胆固醇、高果糖、高蔗糖,以及蛋氨酸-胆碱缺乏饮食诱导、L-氨基酸补充饮食诱导等,可以形成与NASH相似的病理组织如肥胖、炎症、纤维化。这些模型强调了动物模型中肝脏甘油三酯积累的潜力,但在病理生理学上却无法模拟临床NASH;其他因素,比如环境温度,也可能会影响NASH动物模型的疾病程度。
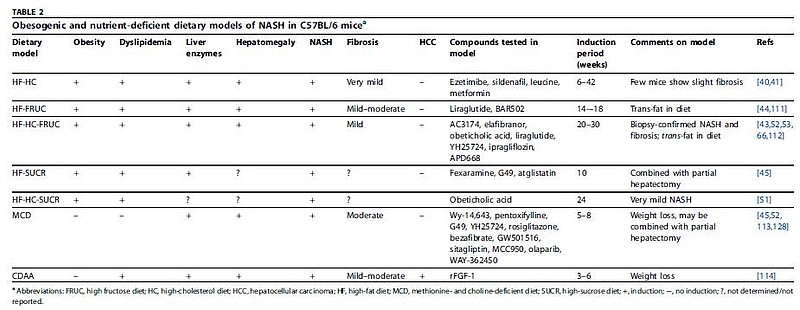
其次是利用化学毒素诱导的模型,利用四氯化碳、硫代乙酰胺、链脲佐菌素等化学毒素,在动物体内诱发肝脏氧化应激反应,导致有害脂质和蛋白质过氧化物不断累积,并发生严重的坏死反应,从而使肝细胞结构和功能被破坏,模拟肝纤维化或肝硬化等形态。然而这些模型发病机制、病程变化及组织学形态与人类NASH差异较大,并且药物毒性强,易致动物死亡。
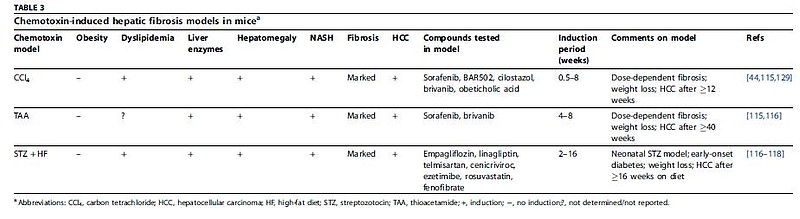
然后是基因突变模型,对动物的基因进行人为改造,来模拟各种不同突变因素的动物模型,例如ob/ob纯合突变小鼠会发生肥胖和脂肪变性、db/db小鼠纯合突变会导致瘦素受体失能等。但这些模型往往无法完整模拟整个NASH疾病进展过程,需要饮食或化学物质诱导进行“二次打击”来最终达到NASH表型。
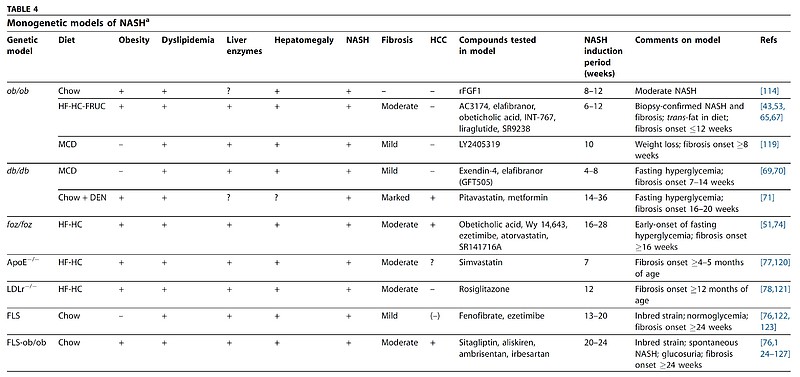
最后是复合模型,前面三种模型均不能完全模拟人类NAFLD的发病机制,其表型与人类NAFLD存在差异,往往会把基因编辑模型与饮食或药物诱导相结合产生复合模型,使复合模型的表型和发病机制与人类NAFLD更接近,并能反映疾病从单纯性脂肪肝向NASH进展,NASH向肝纤维化进展的过程。
2.2.2. 现有临床阶段药物使用动物模型
不难看出其实目前已经做到临床阶段的药物在临床前选择的动物模型可谓五花八门,基本上交叉都很少,一方面可能是对致病机理的理解各不相同,另一方面也是因为各个药物靶点和试图解决NASH的切入点各有侧重。
以临床进展最为领先的OCA为例,它在临床前试验选取了四种动物模型:C57BL/6老鼠高脂高胆固醇高果糖饲养模型(稳定构建NAS>5),C57BL/6老鼠高脂高胆固醇高蔗糖饲养模型(NAS较低),ob/ob老鼠高脂高胆固醇高蔗糖饲养模型(纤维化更严重),foz/foz老鼠高脂高胆固醇饲养模型(空腹高血糖),这四种动物模型发展成为NASH的机理如下图。

2.3. 临床设计和监管要求
2.3.1. FDA
2018年12月FDA发布了NASH临床研究指南的草稿,对临床二三期试验的设计提出了较为详细的要求。
2.3.1.1. Phase IIa试验
无创生物标记物,如肝功能指标(AST、ALT)、影像数据(肝脏弹性、肝脏脂肪含量),都可以作为POC试验的临床终点,而病理学结果并不必要,研究者可以根据药物特性决定采取病理/影像/生化等指标来作为入组和有效性标准。
试验周期应视药物作用机理和预期能观察到有效性的时间而定,如果选择病理学临床终点则应留有足够长时间以保证能观察到病理学效果
早期临床应当通过谨慎选择入排标准与III期临床计划针对的患者群体保持一致。
2.3.1.2. Phase IIb试验
Phase IIaPOC试验证明药物的药理学有效性,在Phase IIb试验中应以病理学结果为临床终点探索药物的有效性(即炎症减退和/或纤维化改善),为三期临床的剂量选择和统计学分析提供足够数据依据。因需要进行肝穿,试验周期通常至少为12-18个月。
研究者需要对所采用的生物标记物提供依据,如生物标记物对NASH病理学结果的预测能力、以提升入组筛选成功率,又如生物标记物对肝硬化和肝脏相关并发症的预测能力等。
研究者可以采用创新的Phase II/III期结合设计,即在剂量探索阶段完成后紧接着用最终选定的剂量继续试验,在试验开始前需与FDA讨论方案设计和统计学方法。
2.3.1.3. Phase III试验
入组标准包括患者需在入组前6个月内接受过肝穿病理检查,若肝穿距离入组3个月以上需要保证体重基本稳定,NAS评分≥4(炎症和气球样变各≥1)且纤维化评分在1-4之间,基线的末期肝病(End Stage Liver Disease,MELD)评分≤12,伴发有二型糖尿病患者入组前需接受至少3个月药物治疗且病情基本可控,已使用维生素E或吡格列酮的患者入组前需停药或保持6-12个月稳定剂量并做好患者分层。
排除标准包括患者不应有可导致慢性肝病的其他因素(酒精性肝病、病毒性肝炎、PBC、PSC等),建议排除胆红素大于等于1.3mg/dL和国际标准化比值(International Normalized Ratio,INR)大于等于1.3的患者、门静脉高压患者、ALT和AST水平超出正常上限5倍以上的患者。
Phase III试验应为双盲安慰剂对照,最终目标是减缓、停止或逆转NASH的疾病进程以及改善临床结果(包括防止进展到肝硬化、减少肝移植需求、改善生存率),FDA建议研究者采用肝穿病理结果的改善作为临床终点、用于预测临床获益来加速审批,有效性终点应设置为以下三者之一:①脂肪性肝炎消退(定义为没有脂肪肝病或只有孤立简单的脂肪变性而没有脂肪性肝炎、NAS中炎症评分为0-1且气球样变评分为0),并且肝纤维化没有恶化;②大于或等于一个级别的肝纤维化改善(NASH CRN纤维化评分),并且脂肪性肝炎没有恶化(定义为NAS评分的气球样变、炎症或脂肪变性没有增加);③同时达到脂肪性肝炎消退和肝纤维化改善,即①+②。
对于以肝穿病理结果为基础获得加速审批的药物,在提交上市申请时需要启动随机双盲安慰剂对照临床试验来验证药物的临床获益,复合临床终点包括延缓以下疾病进展:①在病理学上进展到肝硬化;②肝脏失代偿事件(如肝性脑病、静脉曲张出血、腹水等);③MELD评分从小于或等于12增加到大于15;④肝移植;⑤全因素死亡。
FDA鼓励研究者提出能最终替代肝穿的生化指标或无创影像指标,一旦被FDA认可,研究者可以在患者筛选和有效性评估中使用该等指标。
2.3.2. CFDA
2019年3月CFDA紧跟着发布了NASH治疗药物临床试验技术指导原则(试行),其主要精神可以看出基本上是脱胎于FDA的指南。
2.3.2.1. 临床药理学研究(Phase I)
临床药理学研究包括人体耐受性试验、人体药代动力学和药动学/药效学试验。
由于治疗NASH的药物多需较长时期给药,因此,除非受药物的毒性或药理作用所禁忌,在多次给药耐受性试验中给药的时间应足够长,最少应连续给药一周,若药物及其代谢物血浓度不能达稳态,则应适当延长。
在药物的早期研发阶段,可以考虑以影像学、血清学、NASH肝纤维化无创判别模型作为药效学指标,进行小样本、短疗程的药动学/药效学评估,全面了解药物的暴露/效应作用特点,为后续临床试验提供指导。
2.3.2.2. 早期概念验证临床试验(Phase IIa)
早期概念验证试验应提供初步的证据支持后续的临床试验,包括入组标准、临床试验的周期以及终点的设置。
受试者的入选可采用血清生化检查或影像学方法。主要疗效指标可采用无创性标志物,包括MRI-PDFF、MRE等影像学改变,以及肝脏疾病特异性的血清生化指标的变化等,也可结合组织病理学、影像学和血清生化检查等无创标志物的变化共同评价肝脏脂肪含量和炎症/纤维化。
给药剂量和治疗持续时间要根据作用机制及对于所选疗效指标的预期作用来设计,应有足够长的终点观察时间。建议该阶段包括多个剂量探索作为后续设计的选择。
2.3.2.3. 后期探索性临床试验(Phase IIb)
建议采用安慰剂对照、随机、双盲设计。建议纳入经肝组织病理学确诊的NASH患者,肝组织活检至入组的时间窗一般不超过6个月,在该时间窗内需注意患者是否接受可能影响肝组织学变化的干预。
入组标准包括NAS评分≥4分,其中炎症和气球样变各至少1分;同时,CRN纤维化≥F2。排除标准包括ALT和AST升高超过正常上限(ULN)的5倍,胆红素水平不应超过ULN,MELD评分>12分,CTP评分>5分。
有效性终点应以组织学改善为主要疗效指标,包括①NASH改善同时纤维化无恶化;或②肝组织纤维化改善1分及以上同时NAS评分无升高;或③NASH改善同时肝组织纤维化改善1分及以上。NASH改善定义为NAS评分至少降低2分,其中炎症和气球样变各至少降低1分。这里与FDA指南比较明显的区别是对于NASH改善的定义宽松很多。
可设置多个剂量组,评价药物的量效关系。应保证足够长的研究时间以观察组织学改善,至少要达到12~18月。
2.3.2.4. 确证性临床试验(Phase III)
应设计为随机、双盲、安慰剂平行对照试验,受试者、主要疗效指标、给药剂量和治疗持续时间应与后期探索性临床试验相同。
由于NASH的治疗可能需要长期服药,应包括停药后随访。
在确证性临床试验同时可进行群体药代动力学研究、药物基因组学研究等。
2.3.2.5. 确证临床获益的试验(Phase IV)
鉴于组织学改善的替代终点与临床结局的相关性尚未确立,应进行临床试验确证临床结局的获益,可以接受在上市后开展该试验。