(一)FDA究竟有没有药品上市决定权?
表面上,$信达生物(01801)$ /礼来的信迪利单抗,能不能在美国🇺🇸获批上市?ODAC肿瘤药物审评委员会的专家们才有投票表决的权力,FDA的官们只能躲在一边旁听。
看看去年雪球大V怎么说的?

前天晚上,只要从头到尾观战了那个ODAC所谓的讨论会,你就懂了,这真的是一个美丽的误会。
FDA完全主导了ODAC的风向。FDA的Pazdur和Singh提供了很多不利于礼来的证词,在3期临床入组患者比例达到了80%之时,说ICE没有遵循伦理学原则及时更新《知情同意书》,并特别亮出来证据——时间轴。

在礼来辩解说,私下沟通FDA默许信迪利单抗提交注册之后,Pazdur要求注册申请应该在FDA的正式场合中进行,对的,公事公办,而不是中国人习惯的私下磋商。Singh甚至威胁要公开所有私人信件,礼来那个高管立马反转道歉……
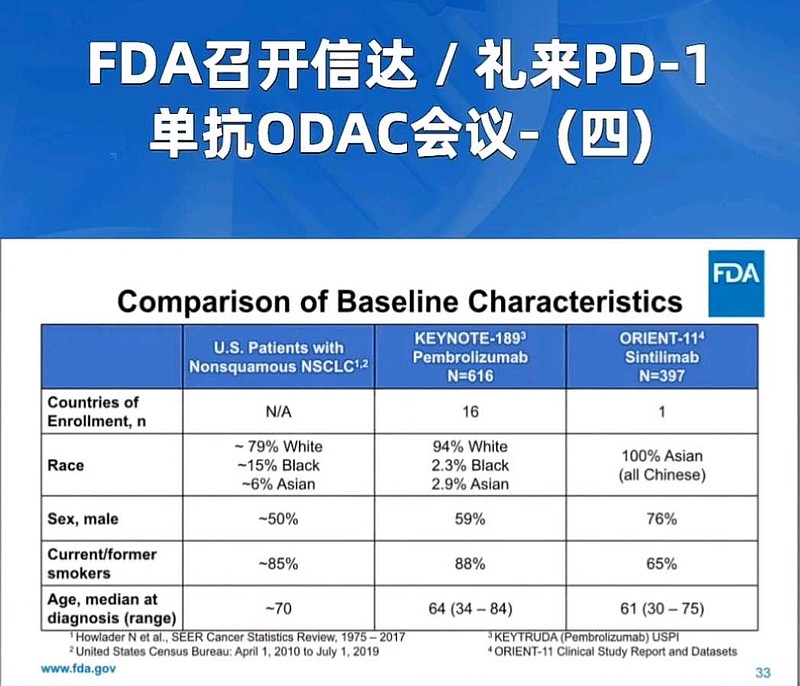
另外,很多朋友认为信迪利单抗不能通过的关键是——人种差异。
不过,FDA显然不是白左,除了Black,给出的差异还包括吸烟率、中位年龄、性别。暗示信达生物选择性招募患者入组。
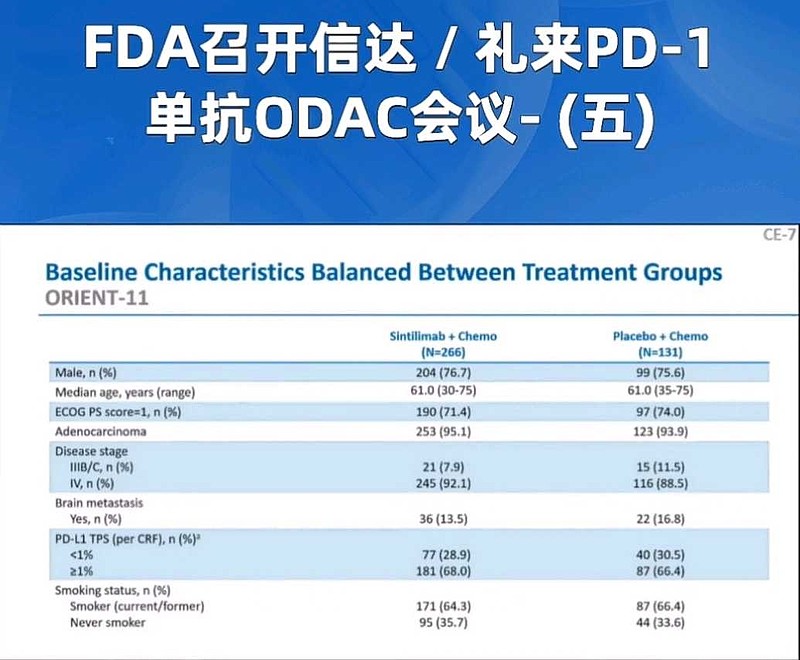
令人费解的是,信达生物提供给ODAC的PD-L1<1%亚组入组比例,仅仅只有28.9%。联想到PD-L1≥50%亚组入组比例高达40%,正常思维的人,都不得不猜测信迪利单抗入组筛选是否有故意嫌疑。
毕竟,PD-1非鳞非小细胞肺癌一线中国3期临床对比,信达生物招募入组患者的PD-L1表达水平也太符合——试验组的利益了吧。
$恒瑞医药(SH600276)$ ,至少PD-L1≥50%亚组入组比例只有14%。
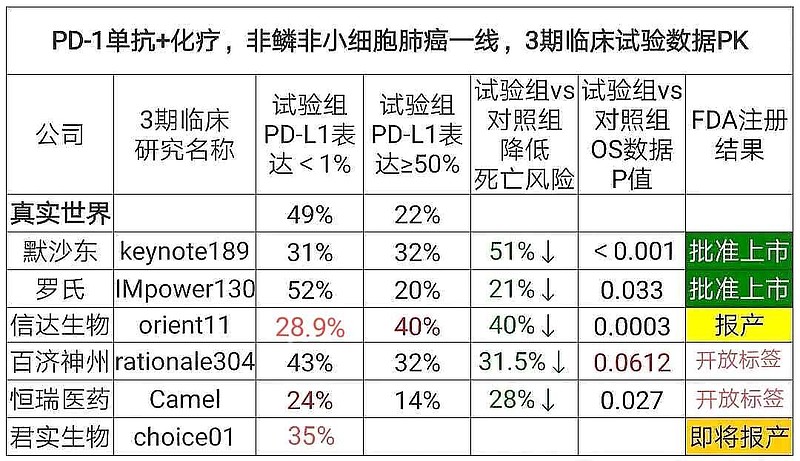
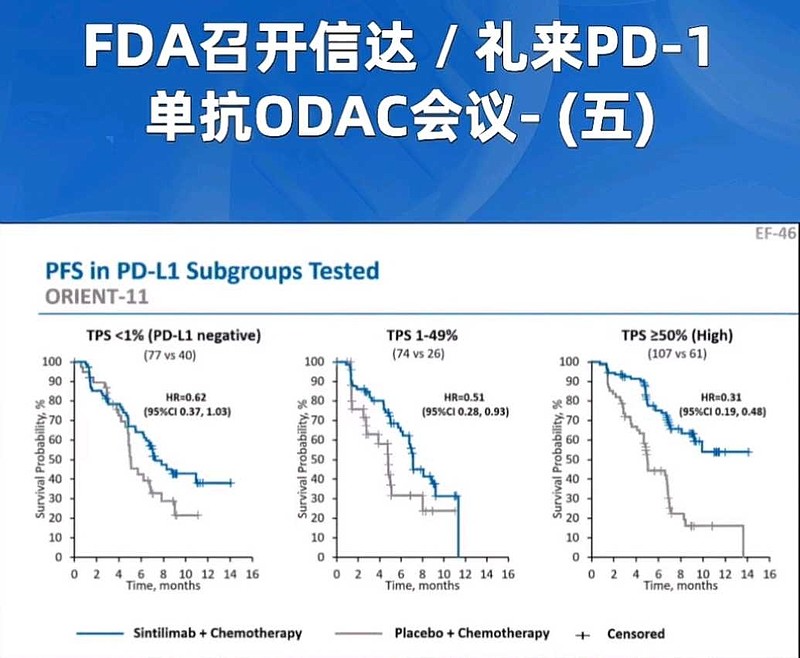
那么,再看看PD-L1<1%亚组降低疾病进展风险是非劣效,PFS HR=0.62(0.37,1.03)。
你就秒懂了,入组筛选对于信迪利单抗疗效有多么重要。
不要以为FDA傻乎乎的看不出来,他们只是看破不说破而已。
西方法律讲究证据,猜测不能作为证据,不能作为呈堂证供。
FDA具有专业水准,当然可以决定药品是否可以上市。
(二)PD-1鼻咽癌临床试验需要黑人入组吗?
市场开始担忧$君实生物-U(SH688180)$ 特瑞普利单抗(PD-1)鼻咽癌适应症,是不是需要黑人入组?是否可以获得FDA批准上市?
FDA给予了君实生物PD-1两项突破性疗法认定,而且不会召开ODAC,直接由FDA审批。难道FDA准备自己打自己的脸,不批准,可能吗?
前面已经说过了,FDA不傻。

Black Lives Matter,黑人的命也是命。肯定是ZZ正确。
那么,亚裔的命,就不是命了?
美国鼻咽癌在亚裔人群高发,也需要得到及时救治。
(三)FDA是不是先礼后兵了?
大家都是文明世界的人,ODAC召开以前,现任肿瘤卓越中心主任的Pazdur,似乎态度陡然突变,要求注册申请应该依据国际多中心研究(MRCTs)。
其实,变化来自于一年前,君实生物中期业绩交流会,就透露了FDA要求PD-1至少30%入组患者来自于欧美国家。
很多信达生物散户不知道罢了。
2月10日FDA的“信达”PD-1专家审评会,引起了中国医药行业很大的反响。像很多事情一样,开会只是择机向公众发声,思想统一和结论商定是会前的事,即功夫是下在会前的。当然,会上的表现也重要,因为FDA不仅要做正确的事,还要让公众看到,justice must be done and be seen done。
先来看FDA会前都做了什么。
当然,只限于公开的信息资料,也就是FDA希望公众了解的东西。
(1)会前一个多月,2021年12月15日,在《新英格兰医学杂志》上,FDA肿瘤卓越中心主任 Pazdur医师和同事发表了题为“西部狂野式的免疫抑制剂研发,The Wild West of Checkpoint Inhibitor Development”的文章。文章指出很多中国药企开发免疫抑制剂类药品,浪费大量资源于无创新意义的药品研发上,并误称这些药为“me better”而不是“me too”。这些用低于美国临床对照标准的方式在中国开发的药,到美国来上市,是不值得FDA给予照顾的。
(2)会前一周,2022年2月4日,在《柳叶刀》杂志上,Pazdur主任又与同事发表了题为“从中国输入临床试验 – 祸水上的桥接,Importing oncology trials from China: a bridge over troubled water”。文章说至少有25个来自中国的免疫抑制剂类新药申请,几乎都是只基于在中国做的临床试验数据,并再次提出FDA是否给予照顾要基于药品的创新性。这里所说的“照顾”,是指regulatory flexibility(监管灵活性)。通常FDA是不允许用单一国家,且是与美国的种族谱很不同的国家的临床试验数据来支持在美国上市申请的。但若是这个药有特殊性,例如:全新类的药品,在美国和其它地区难以入组患者,临床急需等等,就可能给予“照顾”。举例说明包括了鼻咽癌(NPC)和肝细胞癌(HCC)
(3)会议当周,2022年2月7日,Pazdur主任接受Stat媒体的采访,指责中国药企在钻FDA监管政策的漏洞,即原则上允许用美国国外单一国家的临床试验数据来支持上市申请。并强调FDA批药只关注其自身的疗效,不考虑市场竞争因素,针对的是“礼来”号称用廉价中国药来促进竞争、降低药价40%的说法。
《新英格兰医学杂志》和《柳叶刀》均属全球最著名的临床医学杂志,其上发表的文章,即使是政策型而不是研究型文章,也多少要经过匿名同行评议。这些“同行”中不少人的学术声誉、职业稳定性和经济独立性,恐怕都高于FDA官员,所以其评议有一定的客观性。Stat是医药界受尊重的专业媒体,常常对FDA的一些做法提出尖锐的批评意见。
至此,几乎所有人都明白了,信迪利单抗恐怕不能在美国上市了。
但是,信达生物/礼来仍然坚持不撤回上市申请。事后美名其曰‘‘出海闯关第一’’、‘‘以后中国药企都要摸着信达过河’’。
这种阿Q式的精神胜利法,不知道对于信达生物股价管用不管用。
幸亏ODAC不是现场答辩会,仅仅是网络会议。不然,礼来的代表可能要羞愧得钻地缝了。
(四)支持礼来的那一票,是认真的吗?
ODAC主持人反复(一共三次,在不同的会议时间段)强调专家、计划和即席发言人,要说明是否与企业有经济利益关系,并说不在乎是否有关系,但在乎是否公开。
最后的专家投票表决中,14:1,投唯一反对票并当场理直气壮地发表了很长讲话的美国南加州大学Jorge Nivera教授 , 就在会前说明了他的研究得到企业的赞助。他的参会并享有投票权,是在FDA经过审查后专门批准的。他的说明书(3页)和FDA批准他参会并表决的信(4页)都包含在FDA会前24小时公布的材料中。
所以,连这一票都是FDA放出来的。你们还敢于说,FDA没有决定权?
(五)打铁尚需自身硬
FDA说了,默沙东、百时美施贵宝、罗氏、再生元制药都是OS终点。
为什么信达生物你使用PFS终点?而不是使用OS终点?
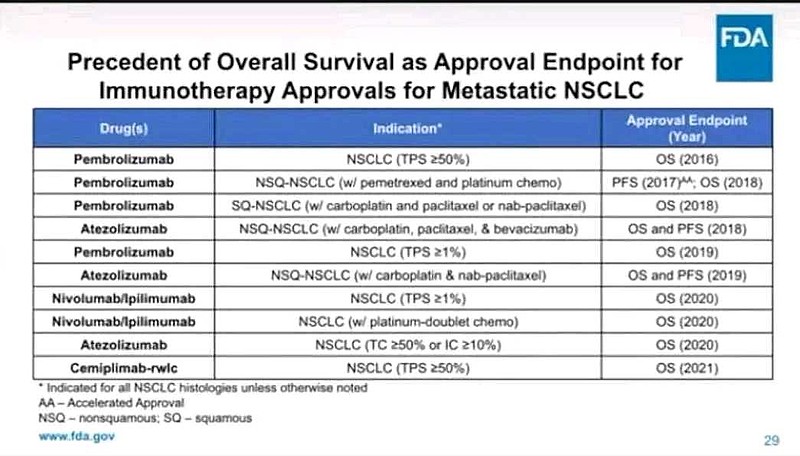
信达生物当然不可能回答,为了抢进度赶工期啰。
小部分信达生物散户鼓吹的执行力强,不会指的是——筛选病例或者用PFS不用OS的能力吧?

人种差异只是借口,临床试验质量才是根本。
(六)中国药企的销售能力,从来不是护城河
中国药企的销售能力从来不是护城河,只有好的产品才是。
临床研发,才是台上三分钟台下十年功的护城河。
总结
信达生物PD-1出海失败的启示,出海需要懂得规则,学会规则,遵守规则。而不是打破规则,去me-too别人家的临床试验设计方案。
君实生物从小适应症开始临床试验,信达生物从大适应症开始临床试验,现在谁的策略更好,已经慢慢显露出来。
不然,FDA把信迪利单抗发回药企去重做3期临床,与默沙东K药头对头对照的时候,你就要叫哭了。
ODAC讨论会上,FDA的要求还是很明确的,相同适应症me-too,基本上都要求跟美国已批准的PD-1做头对头对照研究。
礼来预估,那将是几亿美金和若干年的巨大投资,需要2000人,到2030年才会完成。届时2029年,默沙东K药核心专利已经过期了。
所以,君实生物PD-1反而选择了一条正确的出海之路。unmet medical needs #提供未满足的医疗需求# ,也许道出了FDA内心的真实想法。