医药行业的前几十年是小分子的天下,而随着大分子的崛起,小分子的地位岌岌可危。随着siRNA、ASO等药物上市,越来越多的声音认为,RNA药物是继小分子和大分子之后的第三代药物。让小分子更加失去颜色的,是AAV、CRISPR等基因疗法的兴起,“一针治愈”给大家带来了更多的想象。
有意思的是,在SMA(脊髓性肌肉萎缩症)这个罕见病中,小分子、RNA和基因疗法神奇地相遇了。因此我也很好奇,在这样的场景下,到底谁更好?
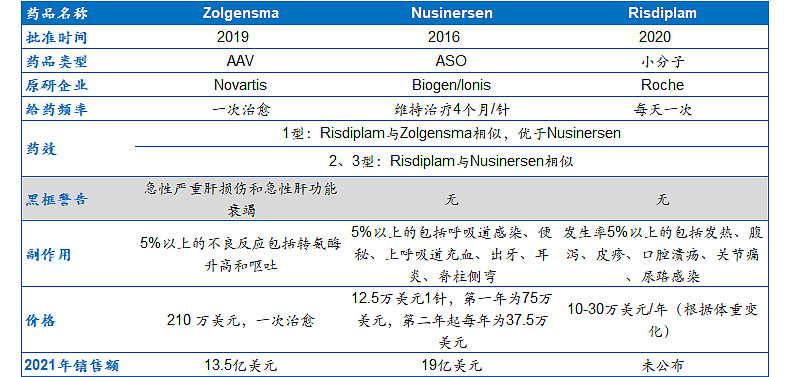
关于各种基本信息好找,而最关键的是疗效的信息,由于没有头对头的试验,很难直接比较。而刚好有一篇meta分析(如最后参考文献),比较了这三种药物的疗效和安全性。
上面文章的结论是,对于1型患者,Risdiplam与Zolgensma的疗效和安全性相似,Risdiplam优于Nusinersen;对于2型和3型患者,Risdiplam与Nusinersen的疗效和安全性相似,但没有Zolgensma的数据。
下面引用文章的介绍:
脊髓性肌萎缩症 (SMA) 是一种常染色体隐性遗传的神经肌肉疾病,每 10,000 名活产儿中的发病率约为 1 。SMA 的特点是进行性运动神经元退化和肌肉无力,这是由于SMN1基因纯合缺失或功能丧失突变导致的 SMN 蛋白水平不足引起的。旁系同源基因SMN2能够指导低水平功能性 SMN 蛋白的产生,但无法完全弥补SMN1的缺乏。
SMA 包含广泛的疾病连续体,分为五种类型(0-4 型),按达到的最大运动里程碑和发病年龄分类 。0 型是最严重的类型,产前发病:婴儿从出生就需要呼吸支持,很少能存活超过 1 年 。患有 1 型 SMA 的婴儿在 6 个月大之前出现临床症状,表现为严重的肌张力减退和虚弱。如果不进行治疗,患有 1 型 SMA 的婴儿无法达到更高的运动里程碑,例如独立坐姿,并且通常在 2 岁之前死亡。
迟发性 SMA(2-4 型)是具有不同表型的异质人群。2 型 SMA 的个体在 7 至 18 个月大时会出现症状 。他们获得独立坐姿,但从不独立行走。预期寿命缩短,52% 的人活到 40 岁 。在 3 型 SMA 中,症状发作发生在 18 个月后 。个人的预期寿命几乎正常,可以独立行走,但随着时间的推移可能会失去这种能力。在 4 型 SMA(最不严重的 SMA)中,症状出现在生命后期(10-30 岁)。个人能够行走,但在以后的生活中可能需要助行器。
关于这三个药物的机理:
SMA由SMN基因调节,SMN1缺失则会引起SMN不足,而SMN2基因和SMN1的差别在于,SMN2由于缺少外显子7,只能产生更短、更不稳定的SMN。
Zolgensma是SMN1基因替代疗法,SMN1基因是SMN蛋白生产的主力军,这种疗法可以最大程度的恢复SMN蛋白活力。
Nusinersen的作用机制是,与SMN2的pre mRNA结合(占位作用),阻断了导致外显子7被排除的剪接信号,从而使得SMN2也包含外显子7,表达正常的SMN蛋白。
Risdiplam的作用机理与ASO的Nusinersen类似,也是调节mRNA剪切的。
meta分析文章:
Ribero VA, Daigl M, Martí Y, Gorni K, Evans R, Scott DA, Mahajan A, Abrams KR, Hawkins N. How does risdiplam compare with other treatments for Types 1-3 spinal muscular atrophy: a systematic literature review and indirect treatment comparison. J Comp Eff Res. 2022 Apr;11(5):347-370. doi: 10.2217/cer-2021-0216. Epub 2022 Jan 18. PMID: 35040693.