撰文丨李杰,勤浩医药首席科学官
编译丨不器
编辑丨于靖
如果要阻挡扬子江的水流,在上游建坝更有效,还是在下游建坝更有效?这个问题没有实际意义。在任何地方筑坝都会使水流停止,无论时间多么短暂。
那么生化级联呢?阻挡上游目标更有效,还是阻挡下游目标更有效?这正是我们在研究细胞外信号调节激酶-1/2(ERK-1/2)抑制剂时遇到的问题。

01 上游还是下游?这是一个问题
在众多人类癌症中,Ras/Raf/MEK/ERK(丝裂原活化蛋白激酶,MAPK)信号转导通路被广泛激活。而药物发现方面,MAPK级联可谓硕果累累。在该通路的上游,目前已有两种不可逆的共价KRASG12C抑制剂获得FDA批准:安进的sotorasib(Lumakras)和Mirati的adagrasib(Krazati)。
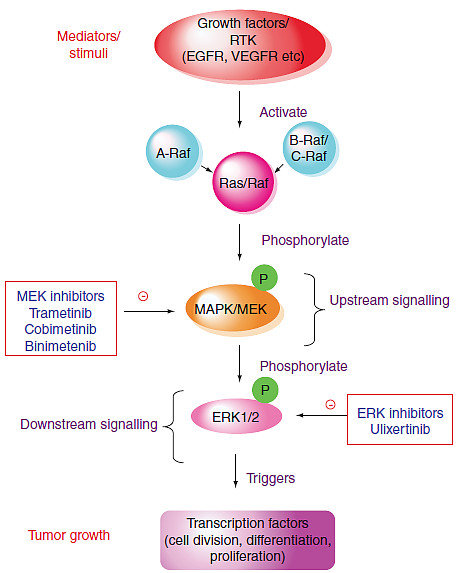
同时,自2011年Plexxikon的vemurafenib(Zelboraf)出现以来,B-Raf抑制剂也在激增。GSK的达拉非尼(Tafinlar,2013年)和Array BioPharma的安戈非尼(Braftovi,2018年)也加入了B-Raf抑制剂的行列。
MEK紧邻ERK的上游。MEK抑制剂包括GSK的曲美替尼(Mekinist,2013年)、Exelixis的cobimetinib(Cotellic,2015年),以及Array BioPharma的binimetinib(Mektovi,2018年)。实际上,这些MEK抑制剂在所有激酶抑制剂中是相当独特的。它们不仅是异构抑制剂,现在还被发现具有分子胶的功能。
与KRAS/B-Raf和MEK抑制剂相比,ERK1/2小分子抑制剂的开发相对滞后,迄今为止尚未有ERK1/2获批上市。
但阻断ERK1/2酶的理由再充分不过了。一旦被激活,ERK1/2会使50多种下游底物的丝氨酸/苏氨酸残基磷酸化,并激活细胞膜和核蛋白,从而促进细胞生长和增殖。事实上,pERK水平是衡量所有激酶抑制剂药效的一个指标。
鉴于ERK1/2在细胞基本过程(包括细胞增殖、生长和存活)中的重要性,ERK1/2在肿瘤适应症方面受到了密切关注,并产生了多个临床候选药物。
02 可逆性ERK1/2抑制剂
2009年,Vertex披露了一系列新型嘧啶吡咯ERK抑制剂。其中,VEX-11e(1)是ERK2的强效粘合剂,口服生物利用度高[1]。它的右侧部分已成为一种持久的药源性,在随后的许多可逆性ERK1/2抑制剂中都能见到。
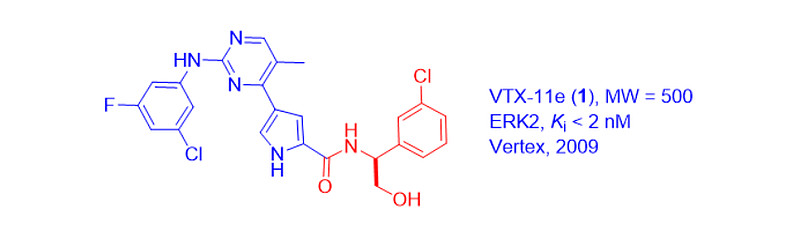
当然,Astex的ASTX029(2)也含有类似的药理作用。它能调节ERK1/2的磷酸化和催化活性,目前正在晚期实体恶性肿瘤患者中进行I/II期临床试验[2]。
从Astex的经验中,我们可以学到药物设计方面的重要一课。
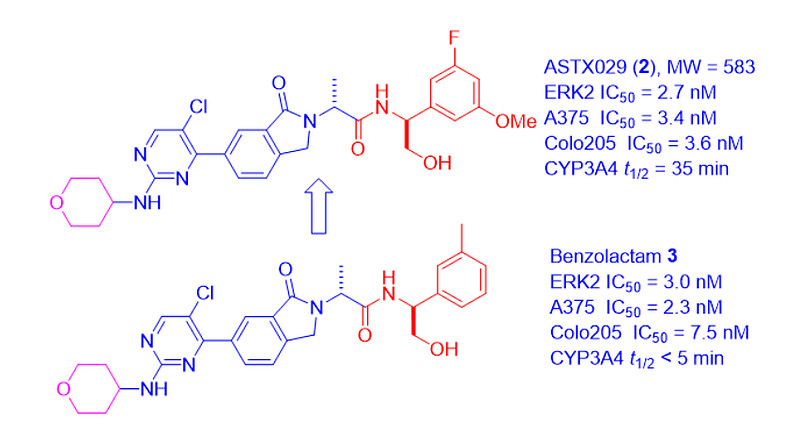
作为基于片段的药物设计(FBDD)的先驱,Astex通过X射线晶体学和生物物理片段筛选,并在结构指导下进行优化,成功地将苯唑内酰胺3确定为一种强效的ERK1/2抑制剂,并从铰链生长到口袋[3]。
苯唑内酰胺3是一种高效的ERK1/2抑制剂,具有优异的激酶组选择性,但其半衰期短,不到5分钟。它主要的缺点是右侧的甲苯基团和左侧的氧杂环(四氢吡喃,THP)。神奇的是,用1-氟-3-甲氧基苯基取代苯唑内酰胺3上的甲苯基,得到了在CYP3A4中半衰期更长(35分钟)、总体药代动力学特征和口服生物利用度良好的ASTX029(2)[2]。
有趣的是,固定苯唑内酰胺3左侧的软肋似乎同时也将其左侧氧杂环的代谢降至最低。
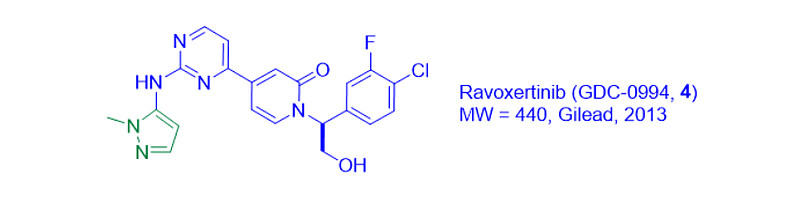
吉利德发现的ravoxertinib(GDC-0994,4),也为药物代谢最小化提供了可资借鉴的经验[4]。
吉利德与Array BioPharma合作,发现了一系列新型ATP竞争性ERK1/2抑制剂,哌啶嘧啶脲5就是其中的代表。
尽管脲5具有强效和良好的体外ADME特性,但在多个物种(包括小鼠、大鼠、小猎犬和金丝猴)中,存在清除率高和口服生物利用度低的问题。科学家通过“骨架跃迁”(Scaffold-hopping)发现了吡啶酮6,它是一种强效、选择性和高效的ERK1/2抑制剂[5]。
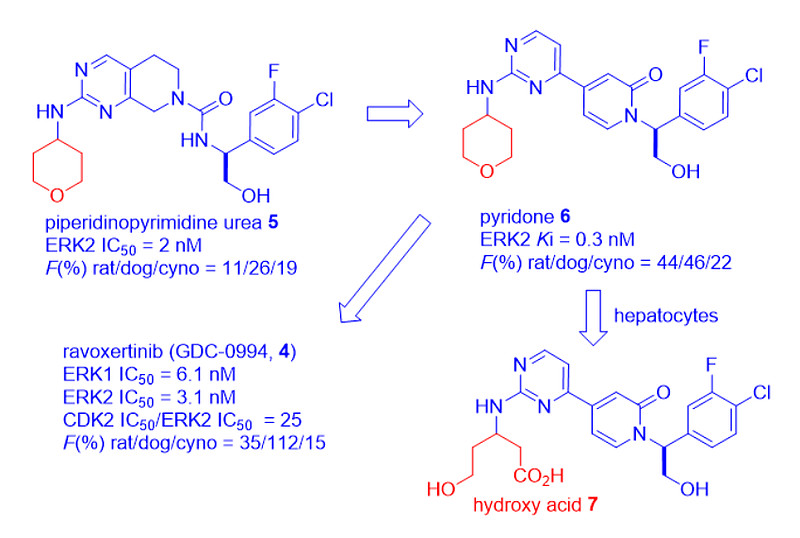
遗憾的是,尽管吡啶酮6具有许多理想的属性,但根据小鼠异种移植数据的PK/PD建模和异计量比例推算出的人体剂量,被认为过高(> 1克/天)而令人无法接受。
将吡啶酮6在小鼠、狗、大鼠和金丝猴肝细胞中孵育3小时后,鉴定出羟基酸7为主要代谢产物。用杂芳族取代软THP分子的努力,最终导致N-甲基吡唑成为最佳选择。由此产生的ravoxertinib(4)显示出优异的临床前PK/PD和药效特征。加上耐受性研究中相对安全的特征,ravoxertinib(4)被转入临床试验[4]。目前,该药正处于I期临床试验阶段,可单独或与MEK抑制剂cobimetinib联合用药。
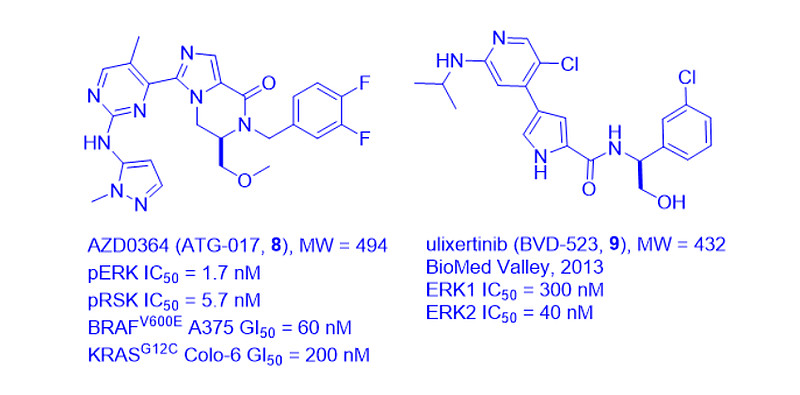
在ERK1/2抑制剂中,N-甲基吡唑取代基显然对CYP3A4代谢具有抗性。它还出现在阿斯利康的AZD0364(ATG-017, 8)中,这是一种强效、选择性的ERK1/2口服抑制剂[6]。
此外,BioMed Valley和Vertex于2017年披露了其ERK1/2抑制剂候选药物ulixertinib(BVD-523,9)[7]。据今年的报道,Ulixertinib(9)是首个同类ERK抑制剂,在MAPK驱动的临床前小儿低级别胶质瘤模型中显示出良好的活性[8]。
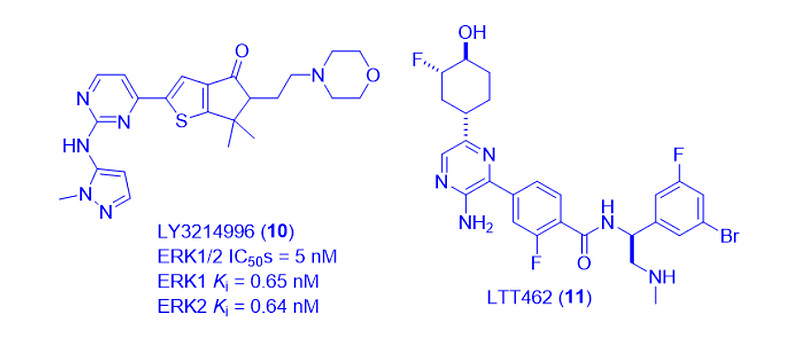
其他几种ERK1/2抑制剂,目前也在临床试验中。礼来的LY3214996(10,II期)[9]和诺华的LTT462(11,Ib/IIa期)[10],就是另外两个例子。
2022年,业内发表了一篇关于ERK1/2抑制剂的综述[11]。
03 不可逆共价ERK1/2抑制剂
靶向共价抑制剂正备受关注,成为流行。
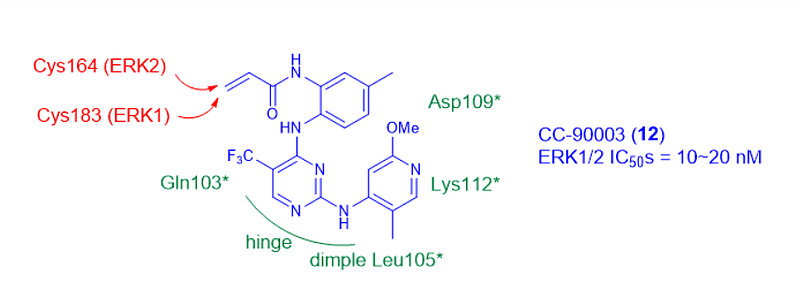
Celgene/BMS的不可逆共价ERK1/2抑制剂CC-90003(12)在复发或难治性BRAF或RAS突变肿瘤患者中进行了Ia期临床试验[12]。
Astex于2015年披露了他们的不可逆共价ERK1/2抑制剂13。共价抑制剂13从高通量筛选的苗头(hit)开始,被发现作为一种选择性共价工具化合物,用于潜在的体外和体内研究,以评估靶向ERK1/2的风险、益处[13]。
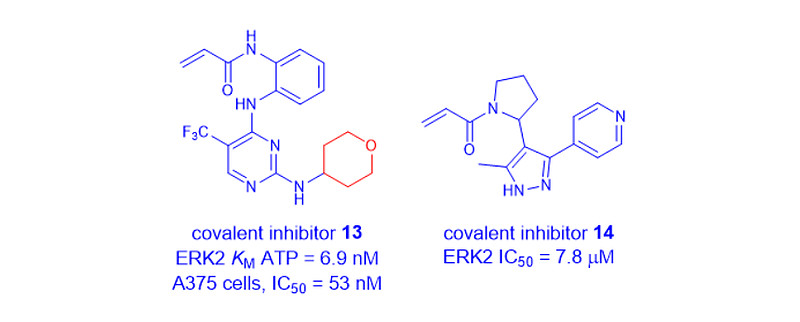
2022年,Astex利用高通量蛋白质晶体学作为主要的靶点发现技术,设计和表征了一个以丙烯酰胺为重点的亲电片段库,并针对ERK2进行了筛选。该团队确定了一种共价ATP竞争性抑制剂14,它与ATP催化口袋中的Cys166残基共价结合[14]。
04 异构ERK1/2抑制剂
异构抑制剂相对于正构抑制剂的优势是众所周知的。
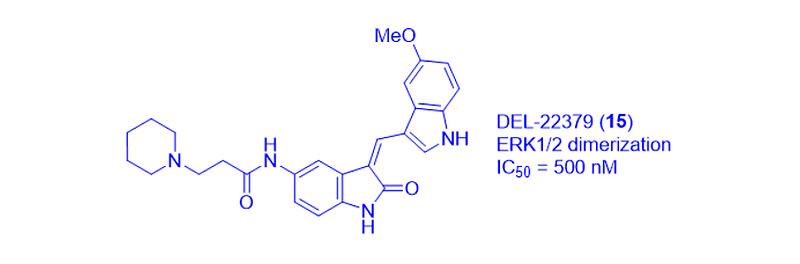
早在2015年,Herrero等就报道了一种小分子抑制剂DEL-22379(15),它可以抑制ERK1/2二聚化,IC50为500 nM。它不改变ERK磷酸化,因此对ERK激酶活性没有影响。DEL-22379(15)的结合位点被确定在ERK2二聚化界面的沟槽中。据推测,15通过对接二聚化界面抑制ERK二聚化,从而阻止两个ERK分子之间的相互作用[15]。
2019年,Sammons及其同事公开了一系列化合物,这些化合物能与ERK2的D-招募位点(DRS)结合,从而抑制ERK2磷酸化DRS靶向底物。此外,该化合物能以剂量依赖的方式阻断组成型活性MEK1突变体对ERK2的激活,IC50为9.9 μM。遗憾的是,这些化合物并不像药物,只能作为很好的工具化合物[16]。
05 总结
从我们开始研究ERK1/2到现在已经过去了二十多年。尽管已有十几种ERK1/2抑制剂进入临床试验,但迄今为止还没有一种获得FDA批准。
其中,Vertex的ulixertinib(9)在MAPK驱动的临床前小儿低级别胶质瘤模型中,显示出了良好的活性。
谁能首先发现ERK1/2抑制剂在更多患者中的疗效,谁就将成为ERK领域的最大赢家。
参考文献:
1.Aronov, A. M.; et al. Structure-guided design of potent and selective pyrimidylpyrrole inhibitors of extracellular signal-regulated kinase (ERK) using conformational control. J. Med. Chem. 2009, 52, 6362–6368.
2.Murray, C. W.; Berdini, V.; Bevan, L.; Buck, I. M.; Carr, M. G.; Courtin, A.; Coyle, J. E.; Day, J. E. H.; East, C.; Fazal, L.; et al. Discovery of ASTX029, A Clinical Candidate Which Modulates the Phosphorylation and Catalytic Activity of ERK1/2. J. Med. Chem. 2021, 64, 12286–12303.
3.Heightman, T. D.; Berdini, V.; Braithwaite, H.; Buck, I. M.; Cassidy, M.; Castro, J.; Courtin, A.; Day, J. E. H.; East, C.; Fazal, L.; et al. Fragment-based discovery of a potent, orally bioavailable inhibitor that modulates the phosphorylation and catalytic activity of ERK1/2. J. Med. Chem. 2018, 61, 4978‒4992.
4.Blake, J. F.; Burkard, M.; Chan, J.; Chen, H.; Chou, K.-J.; Diaz, D.; Dudley, D. A.; Gaudino. J. J.; Gould, S. E.; Grina, J.; et al. Discovery of (S)-1-(1-(4-Chloro-3-fluorophenyl)-2-hydroxyethyl)-4-(2-((1-methyl-1H-pyrazol-5-yl)amino)pyrimidin-4-yl)pyridin-2(1H)-one (GDC-0994), an Extracellular Signal-Regulated Kinase 1/2 (ERK1/2) Inhibitor in Early Clinical Development. J. Med. Chem. 2016, 59, 5650‒5660.
5.Ren, L.; Grina, J.; Moreno, D.; Blake, J. F.; Gaudino, J. J.; Garrey, R.; Metcalf, A. T.; Burkard, M.; Martinson, M.; Rasor, K. et al. Discovery of Highly Potent, Selective, and Efficacious Small Molecule Inhibitors of ERK1/2. J. Med. Chem. 2015, 58, 1976‒1991.
6.Ward, R. A.; Anderton, M. J.; Bethel, P.; Breed, J.; Cook, C.; Davies, E. J.; Dobson, A.; Dong, Z.; Fairley, G.; Farrington, P.; et al. Discovery of a Potent and Selective Oral Inhibitor of ERK1/2 (AZD0364) That Is Efficacious in Both Monotherapy and Combination Therapy in Models of Nonsmall Cell Lung Cancer (NSCLC). J. Med. Chem. 2019, 62, 11004‒11018.
7.Germann, U. A.; Furey, B. F.; Markland, W.; Hoover, R. R.; Aronov, A. M.; Roix, J. J.; Hale, M.; Boucher, D. M.; Sorrell, D. A.; Martinez-Botella, G.; et al. Targeting the MAPK Signaling Pathway in Cancer: Promising Preclinical Activity with the Novel Selective ERK1/2 Inhibitor BVD-523 (Ulixertinib). Mol. Cancer Ther. 2017, 6, 2351‒2363.
8.Sigaud, R.; Rösch, L.; Gatzweiler, C.; Benzel, J.; von Soosten, L.; Peterziel, H.; Selt, F.; Najafi, S.; Ayhan, S.; Gerloff, X. F; et al. The first-in-class ERK inhibitor ulixertinib shows promising activity in mitogen-activated protein kinase (MAPK)-driven pediatric low-grade glioma models. Neuro-oncol. 2023, 25, 566‒579.
9.Köhler, J.; Zhao, Y.; Li, J.; Gokhale, P. C.; Tiv, H. L.; Knott, A.R.; Wilkens, M. K.; Soroko, K. M.; Lin, M.; Ambrogio, C.; et al. ERK Inhibitor LY3214996-Based Treatment Strategies for RASDriven Lung Cancer. Mol. Cancer Ther. 2021, 20, 641−654.
10.Li, L.; Huang, M.; Li, L.; Chen, Y.; Tang, S.; Su, Y.; Dong, R.; Ding, J.; Geng, M. Abstract 3760: HH2710, a highly potent and selective erk1/2 inhibitor for the treatment of mapk mutant tumors. Cancer Res. 2020, 80 (16_Supplement), 3760−3760.
11.Fu, L.; Chen, S.; He, G.; Chen, Y.; Liu, B. Targeting Extracellular Signal-Regulated Protein Kinase 1/2 (ERK1/2) in Cancer: An Update on Pharmacological Small-Molecule Inhibitors. J. Med. Chem. 2022, 65, 13561‒13573.
12.Aronchik, I.; Dai, Y.; Labenski, M.; Barnes, C.; Jones, T.; Qiao, L.; Beebe, L.; Malek, M.; Elis, W.; Shi, T.; Mavrommatis, K.; Bray, G. L.; Filvaroff, E. H. Efficacy of a Covalent ERK1/2 Inhibitor, CC-90003, in KRAS-Mutant Cancer Models Reveals Novel Mechanisms of Response and Resistance. Mol. Cancer Res. 2019, 17, 642−654.
13.Ward, R. A.; Colclough, N.; Challinor, M.; Debreczeni, J. E.; Eckersley, K.; Fairley, G.; et al. Structure-guided design of highly selective and potent covalent inhibitors of ERK1/2. J. Med. Chem. 2015, 58, 4790‒4801.
14.St. Denis, J. D.; Chessari, G.; Cleasby, A.; Cons, B. D.; Cowan, S.; Dalton, S. E.; East, C.; Murray, C. W.; O'Reilly, M.; Peakman, T.; et al. X‑ray Screening of an Electrophilic Fragment Library and Application toward the Development of a Novel ERK 1/2 Covalent Inhibitor. J. Med. Chem. 2022, 65, 12319‒12333.
15.Herrero, A.; Pinto, A.; Colon-Bolea, P.; Casar, B.; Jones, M.; Agudo-Ibanez, L.; et al. Small molecule inhibition of ERK dimerization prevents tumorigenesis by RAS-ERK pathway oncogenes. Cancer Cell 2015;28:170e82.
16.Sammons, R. M.; Perry, N. A.; Li, Y.; Cho, E. J.; Piserchio, A.; Zamora-Olivares, D. P.; et al. A novel class of common docking domain inhibitors that prevent ERK2 activation and substrate phosphorylation. ACS Chem. Biol. 2019, 14, 1183‒1194.