2024.02.02,CDE举办了第二期“药审云课堂”。以下为本人对苏亚楠老师“《M4 模块一行政文件和药品信息》基本要求及常见问题”的培训内容记录及学习笔记。供同行参考。
笔记:
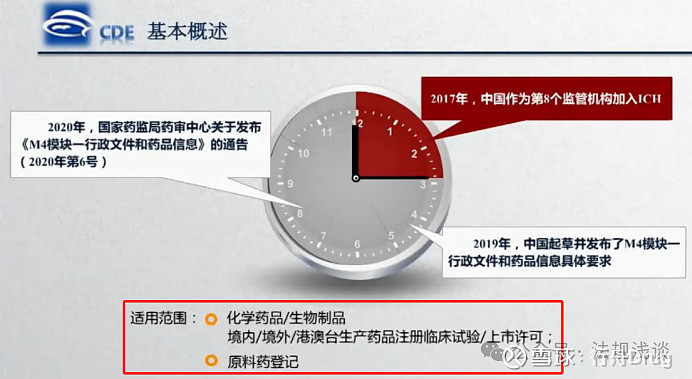
2020年第6号的模块一要求,适用范围包括了化学药品、生物制品的IND和NDA申请,以及化学原料药的登记事项。中药是中国特色,按照《20200927 中药注册分类及申报资料要求》中的要求,有“(一)行政文件和药品信息”的模块,类似模块一。生物制品原液的管理与化学原料药不同,其研究资料与生物制品制剂共同放在CTD资料中,而非登记制。
笔记:
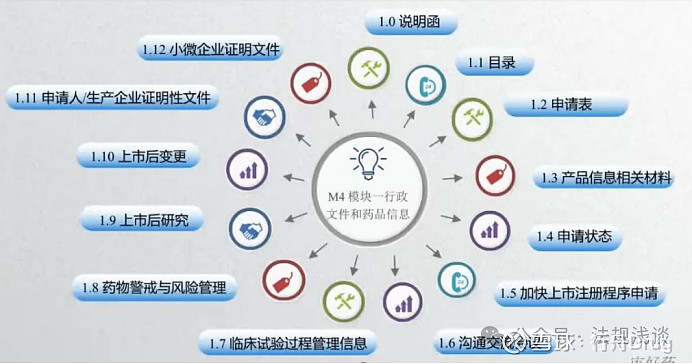
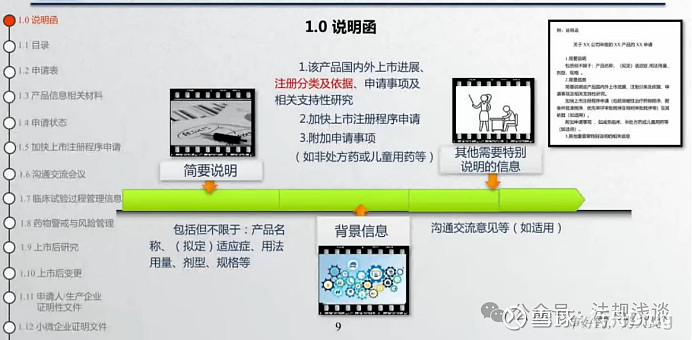
《20200701国家药监局药审中心关于发布《M4模块一行政文件和药品信息》的通告(2020年第6号)》中,没有对1.0说明函的内容做过多详细要求。本次宣讲建议将1.0说明函分以下几个部分:
1.简要说明
包括但不限于:产品名称、(拟定)适应症、用法用量、剂型、规格等
2.背景信息1该产品国内外上市进展、注册分类及依据、申请事项及相关支持性研究2 加快上市注册程序申请3 附加申请事项(如非处方药或儿童用药等)
3.其它需要特别说明的信息沟通交流意见等(如适用)

笔记:
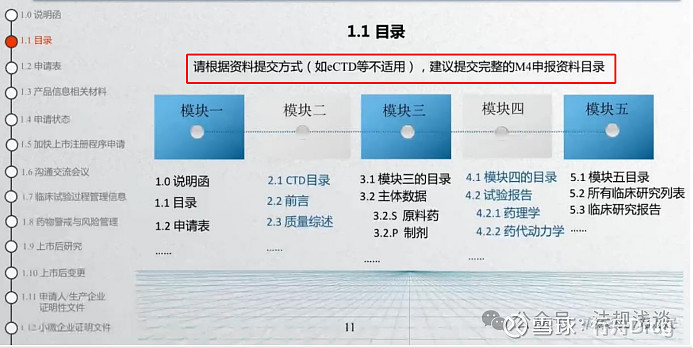
1.1目录为全目录,应“按照不同模块分别提交申报资料目录。”
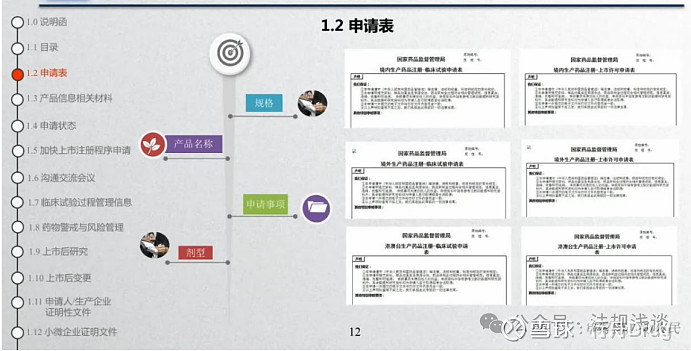


笔记:
原辅包公示平台的三个地址,分别为:登记人(所有权人);生产企业;境内注册代理机构。

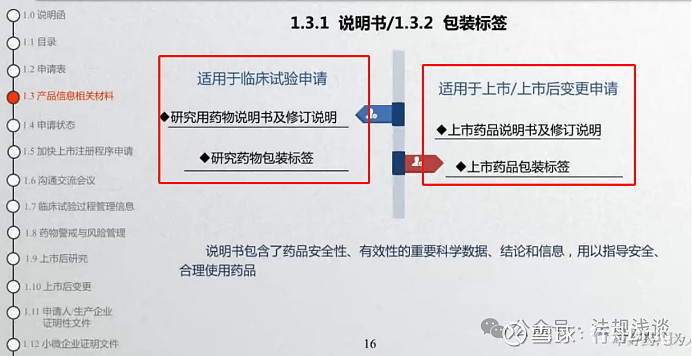
笔记:
1)IND和NDA均需要提供说明书和标签,但因研发所处阶段的不同,说明书的撰写具有阶段性,前者是支持临床研究用(可能很多信息来源于同类药物、同活性成分药物等),后者是支持上市申请/上市后变更申请(必须全面完整)。
2)现行的说明书撰写指南为“20220520化学药品及生物制品说明书通用格式和撰写指南 (2022年第28号)”,但同时还有一系列配套的撰写指南,如“20230320 《化学药品说明书及标签药学相关信息撰写指导原则(试行)》(2023年第20号)”“20240112《抗肿瘤药物说明书安全性信息撰写技术指导原则》”“20210903化学药品和治疗用生物制品说明书中儿童用药相关信息撰写的技术指导原则(试行)”等。
3)关于标签的设计,可参考的指南包括:“20060315药品说明书和标签管理规定(局令第24号)”“20070124关于《药品说明书和标签管理规定》有关问题解释的通知”“T/CNPPA 3024-2023口服药品标签设计指南”等。

笔记:
1.3.3适用于NDA申请,以及上市后补充申请/备案/报告(如适用);笔者理解,在IND时,质量标准及生产工艺/制造检定规程,可提交药物研究过程中的版本。
1.3.4适用于IND申请。

笔记:
因IND和NDA按照适应症管理,因此临床试验方案也应仅针对当前申报适应症;如果同一个适应症涉及了多个临床方案,比如包括食物影响的研究、剂量探索研究、DDI研究等不同临床方案的,应在申请表其它特别申明事项中简要说明。
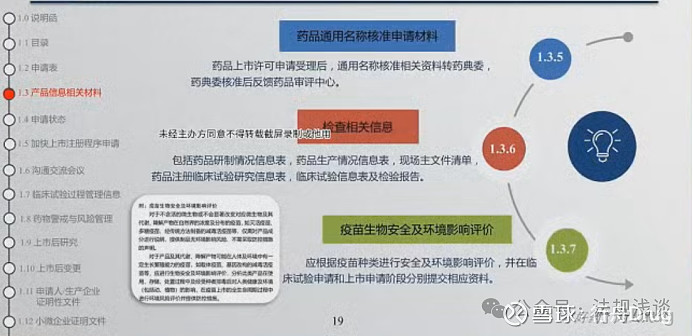
笔记:
1.3.6检查相关信息,适用于上市申请和涉及检查检验的补充申请。

笔记:
2024.03.01后,涉及通用名称核准资料、需非处方药适宜性审查和说明书审核的,不再要求单独提交1套光盘。
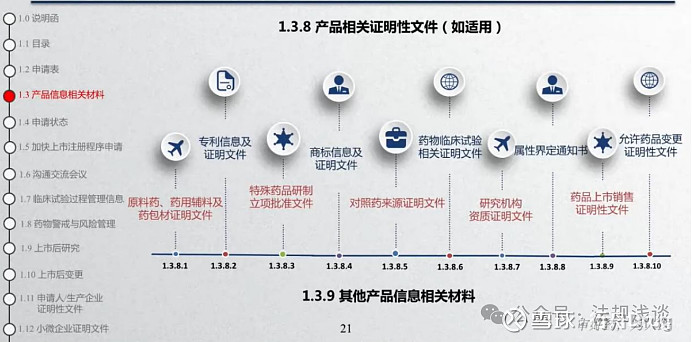
笔记:
1.3.9适用于其它需要特别提供的,与拟申报药品有关的材料。

笔记:
关于原辅包证明性文件的要求:
1)对于制剂未选用已登记原辅包情形:原料药、药用辅料及药包材合法来源证明文件,包括供货协议、发票等。
2)对于制剂选用已登记原辅包情形:原料药、药用辅料及药包材的授权使用书。
另外,对于二次委托的情况,需要附原辅包生产企业对委托供应商的授权信。

笔记:
1)根据《20210704《药品专利纠纷早期解决机制实施办法(试行)》的公告》:“化学仿制药申请人提交药品上市许可申请时,应当对照已在中国上市药品专利信息登记平台公开的专利信息,针对被仿制药每一件相关的药品专利作出声明。”
2)根据《20060315关于进一步规范药品名称管理的通知》:“除新的化学结构、新的活性成份的药物,以及持有化合物专利的药品外,其他品种一律不得使用商品名称”。
3)对于化学药品来说,1、2、5.1类可以申请使用商品名称,3、4、5.2类应选择“不使用”;预防用和治疗用生物制品中1、2类可以申请使用商品名,3类应选择“不使用”;对于按生物制品管理的体外诊断试剂,1类可以申请使用商品名,2类应选择“不使用”。
4)申请商品名的,应当提供商标注册证复印件。关注商标注册证的有效期。
6)如果商标注册证的持有者与注册申请人不一致的,可以由商标注册证的持有者出具商标授权使用协议,或者是官方机构出具的“商标使用许可备案通知书”。

笔记:
在NDA时,应在“1.3.8.6临床试验相关证明性文件”中,对IND批件作业进行逐项答复。


笔记:
1)根据《20200122药品注册管理办法-国家市场监督管理总局令第27号》“第一百二十二条:拟申报注册的药械组合产品,已有同类产品经属性界定为药品的,按照药品进行申报;尚未经属性界定的,申请人应当在申报注册前向国家药品监督管理局申请产品属性界定。属性界定为药品为主的,按照本办法规定的程序进行注册,其中属于医疗器械部分的研究资料由国家药品监督管理局医疗器械技术审评中心作出审评结论后,转交药品审评中心进行综合审评。”
2)1.3.8.8药械组合产品相关证明性文件如属于药品或以药品为主的药械组合产品,应提交药械组合产品的属性界定结果通知书(内部核查)。
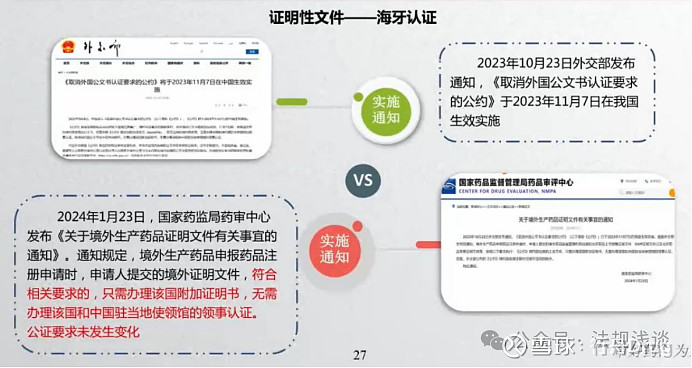
笔记:
根据《20240123 关于境外生产药品证明文件有关事宜的通知》:“《公约》缔约国出具的上述文件,只需办理该国附加证明书,无需办理该国和中国驻当地使领馆的领事认证;但是,外交部公布的《公约》缔约国名单注释中注明不适用的除外。”

笔记:
1)先判断适用范围,再查询缔约国名单,最后提交附加证明书
2)取消了双认证,但公证仍然需要。
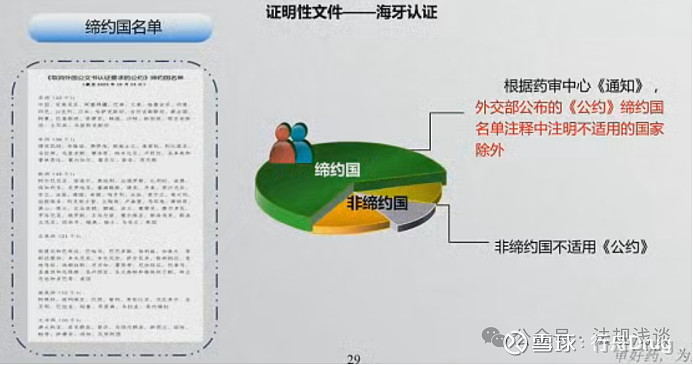
笔记:
关注非缔约国,及不适用公约的国家。
笔记:
根据对应情况提供相应的资料。

笔记:
加快上市注册程序包括:突破性治疗药物程序、附条件批准程序、优先审评审批程序及特别审批程序。
这里附上一张之前CDE培训时的一张PPT,其中清楚地列出了提出阶段及支持政策:
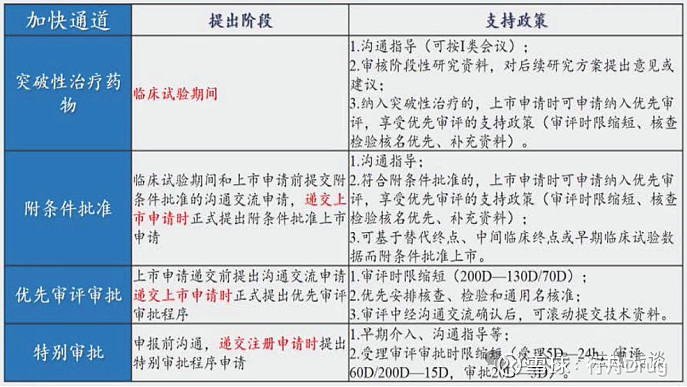

笔记:
1)对于附条件申请,应在临床研究早期提出沟通交流申请,确定附条件批准的关键性临床试验方案,在NDA时正式提出附条件批准申请(申请表勾选);
2)对于优先审评申请,应在NDA申报之前提出沟通交流申请,探讨现有研究数据是否满足药品上市许可审查要求以及是否符合优先审评审批程序纳入条件;在NDA时正式提出优先审评审批申请(在申请表中勾选,同时在“优先审评审批申请系统”提出申请)。


笔记:
1)沟通交流包括三种:依法应沟通交流,原则上应沟通交流,及申请人根据需要的沟通交流。原则上应沟通交流的情形,为pre-IND及pre-NDA之前,这两种沟通交流。pre-三期的沟通交流不属于原则上应沟通交流的情况,当然正常都会去沟通,但其也归属于二类会议。
2)仅需要提交历次沟通交流中,与本次申报有关的沟通交流,无关的不需要提交。

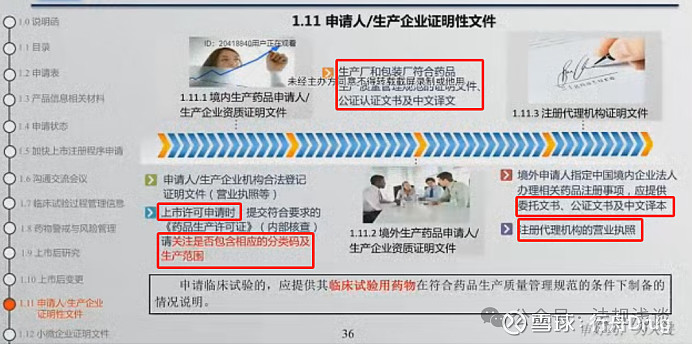

笔记:
1)新增适应症的申请,以及生产企业已获得GMP证明性文件的,仍然需要提交临床试验用药物在符合药品生产质量管理规范的条件下制备的情况说明。
2)法人授权应由法人出具,不能由公司出具;多个法人的,授权签字书应包括所有法人的签字。
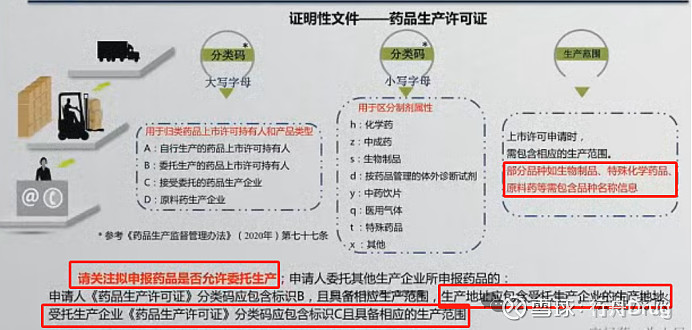
笔记:
1)对于部分品种如生物制品、特殊化学药品、原料药等,药品生产许可证需包含品种名称信息;
2)委托生产的,B证须有相应生产范围,写明受托生产企业的生产地址;C证也应提供,且含相应生产范围。
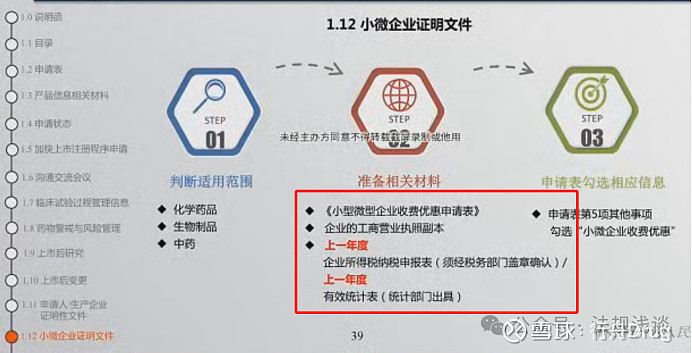
笔记:
1)优惠范围:
符合国务院规定的小微企业提出的符合下列情形的创新药注册申请,免收新药注册费。
1.治疗艾滋病、恶性肿瘤,且未在国内上市销售的从植物、动物、矿物等物质中提取的有效成份及其中药。
2.未在国内外上市销售的通过合成或者半合成的方法制得的化学原料药及其制剂。
3.治疗用生物制品注册分类1类和预防用生物制品注册分类1类。
此外,符合国务院规定的小微企业,已按规定免收临床试验注册费的创新药,在临床试验期间提出的补充申请免收注册费。

笔记:
1)应为上一年度全年的纳税申报表/统计表,不能按季度出具;如,2024年申报的药品,应提交2023年全年的纳税申报表/统计表;
2)很多省市税务系统开放时间在三月上旬,无法提前开具;所以建议尽量避开在一月、二月进行申报,否则无法享受减免。
3)提交临床/上市注册申请须要求,申请人和生产企业同时符合小微企业认定条件。即,申请表中所有申请人均应满足小微企业,包括CDMO企业、生产企业。