CAR-T近年来在血液瘤的治疗中展现出巨大的应用前景,CAR-T包含T细胞激活所需要的第一和第二信号,但目前对CARs和TCRs的差异未能完全阐明,已有研究显示CARs和TCRs之间的一些差异会带来T细胞激活、杀伤和增殖方面的影响。
背景
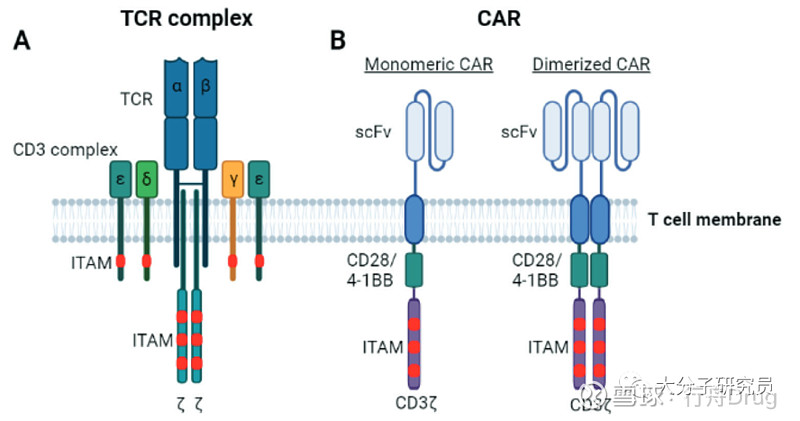
在部分血液瘤中CAR-T疗法以其高缓解率和持久的响应获得极大的关注,目前FDA已经批准4款CD19和2款BCMA靶向的CAR-T疗法。CAR-T是通过基因工程改造在患者的T细胞表面表达嵌合抗原受体(CAR),CAR由三部分构成胞外区、跨膜区和胞内区。胞外区一般是识别肿瘤特异性抗原(TAA)的ScFv,在ScFv和跨膜区之间一般也会加一段柔性的间隔序列用来提高杀伤能力;跨膜区一般来自CD4、CD28或者CD8等不同的跨膜区也会影响CAR-T的杀伤能力;胞内区包含T细胞的激活信号,根据不同的胞内信号对T细胞的激活杀伤以及增殖都有较大的影响。

TCR-T疗法目前尚未获批,不过已经有一些疗法在早期临床中展现出不错的治疗前景。TCR-T是通过TCR识别肿瘤细胞表面的p-MHC复合物,而胞内信号则是利用T细胞原本的激活模块。
CAR-T和TCR-T的异同目前尚无系统的比较,本文通过已有的一些研究来讨论它们在作用机制和功能等方面的差异。
TCRs和CARs对抗原的敏感性比较
TCRs
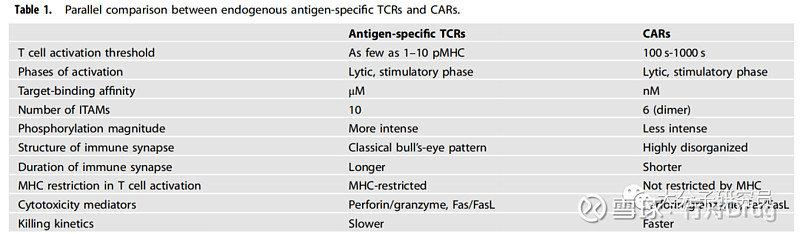
CTLs(抗原特异性T细胞)和靶细胞的相互作用首先是通过黏附分子的非特异性结合然后Ag-TCR(抗原特异性TCR)结合到p-MHC复合物。诱导CTLs杀伤大概需要1-10个p-MHC和数分钟到数小时不等的时间,这跟Ag-TCR和p-MHC结合的亲和力相关,亲和力越低需要结合更多的p-MHC。与仅仅诱导CTLs的杀伤相比完全激活CTLs包括CTLs的杀伤功能、增殖以及因子分泌则需要更多的p-MHC复合物以及更长时间,这说明CTLs的杀伤所需要的阈值更低。有研究将免疫突触(IS)的形态分成“细胞溶解突触”和“细胞刺激突触”。
CARs
与TCRs类似CARs也需要靶细胞表面抗原达到某个水平,一些研究显示诱导CARs杀伤功能所需要的抗原量也远少于完全激活CARs所需要的抗原水平,如诱导CD20 CARs杀伤靶细胞只需要240个CD20分子,而诱导CAR-T增殖和因子分泌则需要5320个CD20分子。这说明CAR-T细胞的杀伤所需要的激活阈值也远小于完全激活CAR-T所需要的阈值。这个具体的数值对于其他的CARs可能没有参考意义,因为CAR-T的激活也受到其他因素的影响,如CAR的表达水平、CAR的亲和力、表位、CAR的长度以及免疫突触的距离等等。
Ag-TCR和CARs的抗原敏感性比较
虽然CARs的设计理念来自TCR和相关共刺激信号并且在组成上也包含了相关的功能域,但是CARS和TCRs的结构差异较大。通常来说,抗体的解离常数为纳摩尔水平,TCR与p-MHC的解离常数为微摩尔水平。有研究显示对于激活T细胞来说,CARs的敏感性弱于TCRs的敏感性,这种敏感性的差异可能由于TCR/CD3复合物激活的胞内信号与CARs激活的胞内信号差异引起。
Ag-TCR和CARs对抗原敏感性差异的原因
ITAM的数量,免疫受体与配体结合后通过胞内ITAM基序的磷酸化来传递信号。TCR-CD3复合物一共包含10个ITAM基序,如此多的ITAM不但可以更快激活T细胞而且可以刺激引起更强的T细胞功能。CAR分子一般只有一个CD3ζ结构域包含3个ITAM基序,当CAR-T细胞激活后形成的CAR二聚体包含6个ITAM基序。更多的ITAM基序对抗原数量的需求越少,TCR信号和T细胞增殖速度与ITAM数量直接相关,但是因子的分泌与ITAM的数量无关。
磷酸化的程度,前面提到TCR可以在较低的抗原密度下激活T细胞,对于信号通路的研究发现经过TCR和CAR刺激后T细胞胞内蛋白的磷酸化水平有较大差异。在T细胞激活的早期,T细胞胞内有282个位点的磷酸化水平差异超过两倍,其中位于CD3δ、CD3γ、CD3ε以及其他信号传导蛋白上的267个位点的磷酸化水平是TCR强于CAR,而CD3ζ和CD28相关蛋白的磷酸化是CAR强于TCR;在更靠后的时间点,基本只有少数的位点存在磷酸化的差异。因此,尽管CAR的设计上包括TCR和共刺激信号,但是CAR并没有完全模拟天然TCR的信号通路。
免疫突触的形态,T细胞和靶细胞之间的相互作用会减慢T细胞的迁移能力以促进免疫突触(IS)的形成,抗原介导的信号传导会诱导T细胞骨架重组以及囊泡向IS的极化,促使更多的Ag-TCR结合到p-MHC复合物并增加相互作用的面积,随着IS附近细胞骨架的重组最终TCR会移动至免疫突触的正中间,在T细胞和靶细胞之间形成有序的类似“靶标”状的结构。

对CAR-T的研究显示免疫突触并不像TCR那样有序,也没能形成“靶标”状的结构,CAR则随机分布在免疫突触中,导致CAR-T细胞无法形成稳定的免疫突触,这也可能是CAR-T下游信号更弱的原因。
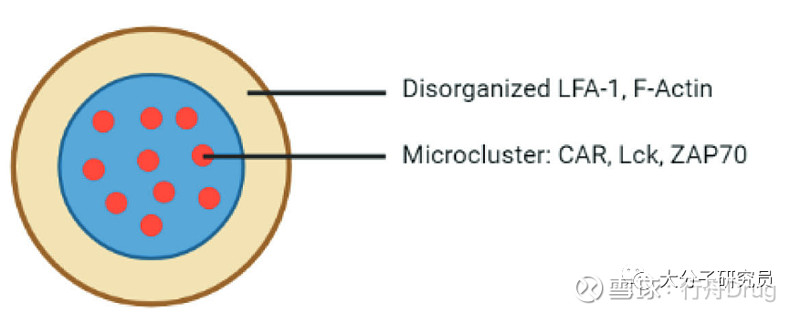
在T细胞激活阶段Ag-TCR和CAR需要不同的抗原密度,尽管Ag-TCR亲和力更低p-MHC密度更小,但是Ag-TCR对T细胞的激活能力更强,这可能是由于TCR具有更多的ITAM基序、更广泛的磷酸化水平和有序的免疫突触共同导致。
TCRs和CARs对T细胞激活能力比较
激活机制
Ag-TCR对T细胞的激活,TCR复合物对T细胞的激活至关重要,TCR结合到p-MHC后通过ITAM的磷酸化传递信号并激活T细胞,在这个过程中T细胞发生极化并在T细胞和靶细胞之间形成“靶标”状的免疫突触,而位于中间的cSMAC区域则是触发TCR信号和分泌细胞毒素的主要位置。研究显示静息状态下CD3复合物的胞内区附着在质膜的内层,当TCR与p-MHC结合后CβFG loop促使CD3结构发生改变暴露出ITAM基序从而开始信号传递。共刺激分子的激活已被证明对于完全激活T细胞至关重要,CD28将Lck招募至免疫突触,由于免疫突触将CD45等排除到IS周边从而解除了对Lck的抑制,Lck将CD3 ITAM磷酸化,随后磷酸化的ITAM招募ZAP70激酶,ZAP70激活后立刻磷酸化下游的底物骨架蛋白LAT,并招募多种下游激酶和信号分子包括Akt,PLCγ等等。
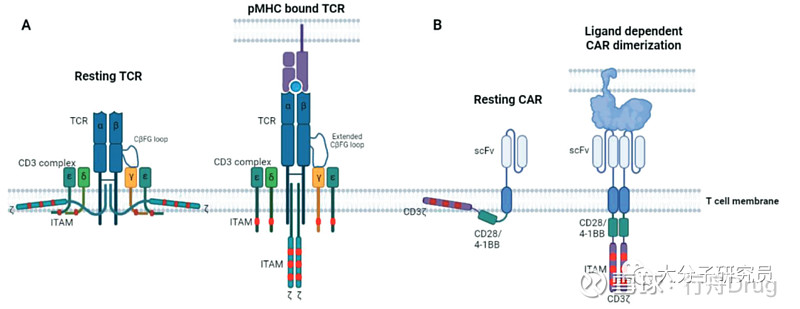
CAR对T细胞的激活,与TCR不同,CAR对靶点的识别并没有MHC限制。CAR可以通过优化自身的结构调整CAR-T的激活和效应功能。例如CAR的跨膜结构域主要来源于CD4、CD28和CD8α,这些结构域对CAR的二聚化以及提高CAR-T的细胞毒性起到重要作用。有研究使用CD3ζ的跨膜区作为CAR的跨膜区,发现CAR可以与内源的CD3ζ以及与其他CAR形成二聚体,这可能是因为CD3ζ跨膜区的半胱氨酸与另一个CD3ζ分子的半胱氨酸形成二硫键所至,如果将二硫键突变掉则会破坏这种相互作用,CAR的功能则被极大降低,需要更多的抗原才能达到相同的激活效果。另外CAR-T和靶细胞之间的IS是无序的,CAR在IS中的分布也是随机的,与TCR相比CAR形成的IS更小也更不稳定。CARs中常用的共刺激信号来自CD28和4-1BB,使用CD28共刺激信号的CAR-T激活更强劲但持久性欠佳,4-1BB则相反激活更温和但更持久。虽然这两种CAR-T背后的机理并不清楚但是可能的原因是使用CD28信号的CAR-T招募更多的Lck。
TCRs和CARs介导的肿瘤杀伤活性背后的机制
细胞毒性的触发,通过前面的比较Ag-TCR仅需要10个p-MHC即可激活T细胞,而CAR则需要数百甚至上千的抗原表达才能激活T细胞。许多研究试图揭示由Ag-TCR和CAR介导的T细胞活化和细胞杀伤的动态过程。

与TCR介导的T细胞活化相比,CAR以更快更强劲的方式激活T细胞,然后更快速地释放细胞毒素。TCR激活的T细胞活化信号持续时间更长,活化的TCR-T细胞和CAR-T细胞具有不同动力学却相似量级的细胞毒素释放。
CAR的IS与由TCR形成的经典IS明显不同,此外CAR IS比TCR IS更小,持续时间更短。LFA-1是一种用于形成经典稳定IS的整合素,但是对于触发T细胞毒性并不是必须的,这也解释了为什么CAR的IS虽弱但能同样杀伤靶细胞。
细胞毒性,T细胞活化会上调细胞毒性囊泡中穿孔素和颗粒酶的表达,之后可以观察到囊泡向IS的cSMAC极化,细胞囊泡和T细胞质膜融合将穿孔素和颗粒酶释放到突触裂隙,在此期间穿孔素在肿瘤细胞上产生孔,然后颗粒酶进入靶细胞诱导细胞凋亡。幼稚T细胞活化后产生数百万个细胞毒性效应T细胞;然而,大多数激活的T细胞因为AICD以及缺乏细胞因子而被清除,剩余的T细胞将其杀伤机制从较快的穿孔素/颗粒酶依赖性途径转变为较慢的Fas/FasL依赖性途径。从肿瘤细胞分离后,T细胞继续以连续杀伤的方式识别其他潜在靶标,穿孔素/颗粒酶介导的细胞杀伤失败会延长IS的持续时间并推迟T细胞解离,这可能诱导T细胞过度刺激以及细胞因子和趋化因子分泌过多;然而,该机制是否与CAR-T细胞治疗相关的毒性,如细胞因子释放综合征和神经毒性,仍有待研究。
CAR与内源性TCR之间的相互作用
将CD19 CAR插入TRAC位点可增强体内肿瘤杀伤
通常CAR-T的构建是通过逆转录病毒载体或者其他随机整合载体在人原代T细胞表面表达CAR,然而随即整合可能导致产生不可预测的CAR-T细胞产物,这促进了在T细胞基因组中选定靶位点特异性插入CAR基因的开发。研究表明在TCRα基因座插入CD19 CAR,观察到CD19 CARs在TRAC-CAR-T细胞上的表达更均一,而通过逆转录病毒转导(RV-CAR)产生的CAR-T细胞虽然CAR的平均表达水平更高但是供体之间的差异也更大。经CD19+靶细胞体外刺激后,TRAC-CAR-T细胞与RV-CAR-T细胞细胞毒性和增殖无显著差异。与RV-CAR-T细胞相比,TRAC-CAR-T细胞发挥了更有效的抗肿瘤活性,据推测这是由于TRAC启动子的转录控制使CAR处于最佳基线表达水平,从而阻止了TRAC-CAR-T细胞的强直信号传导和早期分化。此外,TRAC-CAR-T细胞中表达PD-1、TIM3和LAG3的T细胞百分比远低于常规CAR-T细胞,这与其在体内更优的抗肿瘤功效一致。CAR功能被认为依赖于基线CAR表达水平和受到抗原刺激后CAR的表达改变,这个过程由其在T细胞基因组中的相应启动子调节。
内源性TCR对于CD19 CAR-T细胞在体内的持久性至关重要
T细胞数量不足,化疗导致的T细胞适应性欠佳以及免疫抑制肿瘤微环境仍然是提高CAR-T细胞疗法疗效的挑战。同种异体CAR-T细胞为这些问题提供了潜在的解决方案,但有可能引发移植物抗宿主病(GvHD),从理论上讲,敲除供体T细胞中的内源性TCR可能会阻止受体发生GvHD。一项研究使用CRISPR/Cas9敲除T细胞中编码TCRβ(TRBC)的基因,然后进行CD19 CAR转导,敲除TRBC没有影响CAR-T细胞的活化、增殖、细胞毒性和细胞因子产生的能力,因此敲除内源性TCR可能是预防同种异体CAR-T细胞治疗相关的GvHD而不破坏效应功能的可行策略。然而有研究表明内源性TCR可以增强CAR-T细胞在体内的持久性,从而产生更有效的抗肿瘤活性,因此,敲除T细胞中的内源性TCR可以减轻潜在的GvHD,但代价是体内持久性较短。
CAR和内源性Ag-TCRs都有助于抗肿瘤活性
为了研究CAR的存在是否完全抑制内源性TCR活化T细胞和效应功能,建立了在CD8+ T细胞上表达OT-I特异性TCR和抗HER2 CAR的转基因小鼠,使用OVA257刺激CAR.OT-I和OT.I细胞显示出类似的增殖能力和相同的T细胞激活maker表达水平,说明HER2 CAR没有影响内源性TCR的活性。在同一T细胞上表达TCR和CAR不影响相互的细胞毒性并不一定表明它们的抗肿瘤活性之间是完全独立的,这需要进一步的研究来详细说明内源性Ag-TCRs和CARs在同一T细胞上的相互作用。
CAR-T细胞和工程TCR-T细胞的整体性能比较
过继细胞疗法,包括CAR-T细胞疗法和工程TCR(eTCR)-T细胞疗法,已被证明在治疗癌症方面具有治愈潜力。CAR-T细胞和eTCR-T细胞在不同背景下的整体性能已经有了一定程度的探索。一项研究使用来自原代人CD8+ T细胞构建抗CD20 CAR-T和eTCR-T细胞比较它们被不同抗原表达水平的靶标刺激后细胞毒性、增殖、细胞因子分泌和抑制标志物表达等方面的差异。CD20 CAR-T细胞的杀伤活性无论在低或者高表达CD20的B-ALL细胞存在时均强于eTCR-T细胞,因子表达水平也是 CD20 CAR-T高于eTCR-T细胞。然而,当靶细胞上的CD20密度达到高水平时,CD20 CAR-T细胞的增殖受到抑制,这是CD20 CAR-T细胞PD-1和LAG3表达上调高以及AICD的结果,表明CD20 CAR-T细胞比eTCR-T细胞更容易受到AICD的影响。在长期杀伤测定中,eTCR-T细胞的性能与CD20 CAR-T细胞相当,表明eTCR-T细胞的细胞毒性动力学较慢,这与以前的研究一致。
CAR-T细胞比eTCR-T细胞更容易受到AICD的影响,因为它们具有更强大的活化动力学,这表明eTCR-T细胞的杀伤动力学较慢可能比CAR-T细胞更有益。CAR-T细胞和eTCR-T细胞的功能高度依赖于自身条件以及所处环境,包括抗原密度、CAR或eTCR的表达水平,以及它们对抗原的亲和力等等,这些都需要具体问题具体分析。
结论
在过去的几十年里,一部分液体肿瘤患者,特别是B细胞恶性肿瘤患者,从CAR-T细胞治疗中获益,但大多数接受治疗的患者在输注后仍然复发。TCR-T细胞已被广泛探索,并在临床前环境中取得了显着的成功,并在治疗实体瘤的临床试验中取得了有希望的结果,例如黑色素瘤,肉瘤和肺癌等,这些实体瘤都缺乏独特的表面抗原。CAR和TCR的信号通路与信号调节因子有部分的重叠,但具有不同的磷酸化程度、活化动力学、IS结构和细胞毒性,需要对TCR和CAR进行更深入的研究以提高CAR-T细胞治疗的有效性。