记得本号开刊后的前几篇文章就提到基因治疗(参见【药海听涛】基因治疗是希望还是信仰?),其间表达了我对CGT领域整体的相对悲观态度,尤其是面对腺相关病毒载体的连续出现的安全性问题以及所引发FDA的警惕,使人不由得不为之前景担忧。
在这一年多中,有两款命途多舛的腺相关病毒载体的基因疗法终于获批上市,分别是$拜玛林制药(BMRN)$ 旗下用于治疗A型血友病的BMN-270和$Sarepta疗法(SRPT)$ 旗下用于治疗DMD的SRP-9001,总算是让人觉得基因治疗之路纵然艰辛、但多少还在负重前行。
然而10月31日的一则新闻,好像要击碎这短暂的岁月静好:Sarepta宣布Elevidys(SRP-9001)在三期验证性临床EMBARK研究中,虽然在多个次要临床终点均取得显著性,但未能达到主要终点NSAA。

在这个代号为EMBARK的确证性临床中,共入组125例DMD患者,按1:1分配到Elevidys组和安慰剂组。
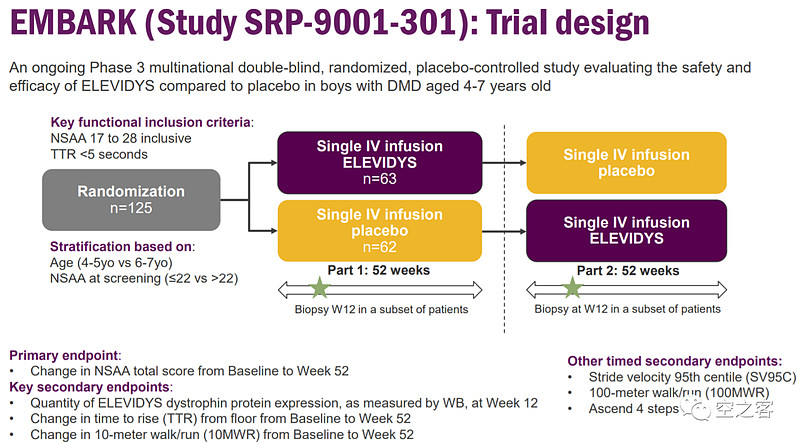
在主要临床终点NSAA上未能达到显著性,而且不是那种差一丢丢的惜败,而是p=0.2441这种毫无翻身余地的失败,4-5岁年龄组也并不显著,6-7岁年龄组更是与安慰剂完全拉不开差距。虽然Sarepta在电话会中反腐强调NSAA终点未能达到的原因是患者随访方式导致,但无论如何总不能理解为什么都没发生。
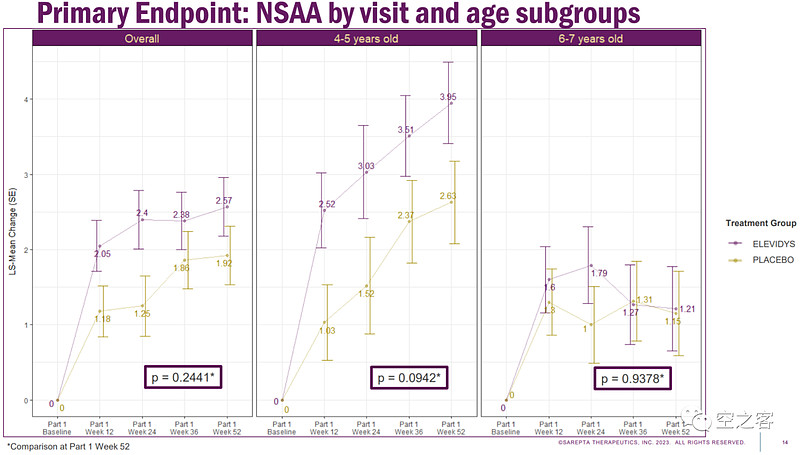
在错失主要终点的情况下,Sarepta只能玩命在次要终点上做文章,确实在TTR(Time to rise)和10MWR(10-meter walk/run)等指标都达到显著性,且公司表示FDA对此结果非常令人鼓舞,并会基于这些次要终点结果争取完整批准。

早在今年6月Elevidys获得FDA加速审批之前,5月12日Adcomm就进行过讨论,最终形成了8:6这样罕见的票型,在会上FDA也很直接的提出了四大方面的担忧,有些已经在后续临床中被验证,对基因治疗或有共性的借鉴意义。

1. 生产工艺和非临床结果
SRP-9001进行过生产工艺变更,而申请用于上市药品的工艺会导致空壳率上升,为什么到上市前会出现这种有点低级CMC的问题我实在也没太明白。


而在临床前研究结果中,小鼠体内micro-Dystrophin的表达和功能都与正常Dystrophin有区别、并为体现出与肌肉比力之间的相关性,大鼠体内micro-Dystrophin的表达也未能有效转化为功能改善、特别是年龄较大的大鼠给药后改善更不明显(这与人体临床结果相符),总而言之就是动物试验很难支持使用micro-Dystrophin作为替代临床终点。



2. 替代终点
SRP-9001的加速审评是基于微小肌营养不良蛋白(micro-Dystrophin)的表达作为替代终点,FDA则怀疑这种生物标志物是否能够预测药物能够带来实际的临床获益。
首先从生物学机制上,SRP-9001表达的micro-Dystrophin,既与正常的Dystrophin有很大差异(缺少多个功能域),又与在症状更温和的BMD(Becker Muscular Dystrophy)患者中表达的短Dystrophin有所不同(缺少蛋白互作域),因而micro-Dystrophin的生物学功能并不明确。

其次,从流病、病理、临床、药理等层面,micro-Dystrophin都缺乏实证依据。

最重要的是临床依据不足,首先临床效果的重要指标NSAA与患者努力程度及临床执行过程都高度相关,且DMD疾病进展又有高度异质性和非线性,这就需要更多的随机双盲对照试验。然而用于支持SRP-9001上市申请的临床研究中,只有一个随机双盲对照试验,即Study 102的第一部分,而其他试验都是开放标签。



对于Sarepta提供的临床结果,FDA也提出了多项疑问:
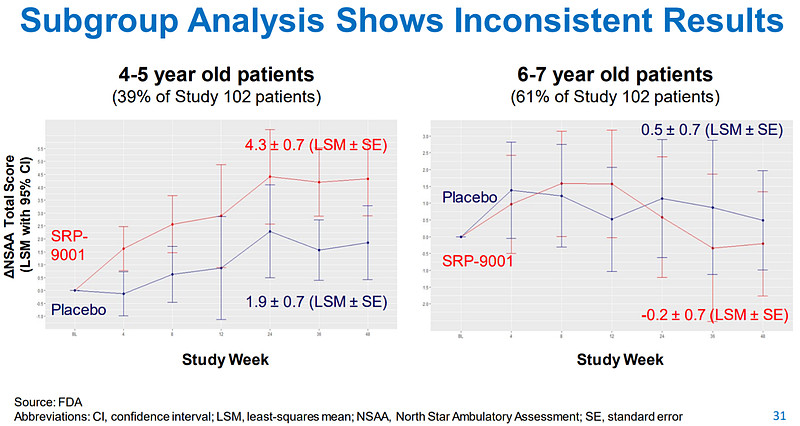
1)在Study 102第一部分中,SRP-9001与安慰剂相比NSAA无显著变化(这也为EMBARK试验在这个主要终点未能达到显著性埋下伏笔),没有清晰的量效关系,不同年龄组之间结果不连续、且6-7岁年龄组药效很差(这也跟EMBARK试验的结果相符)。

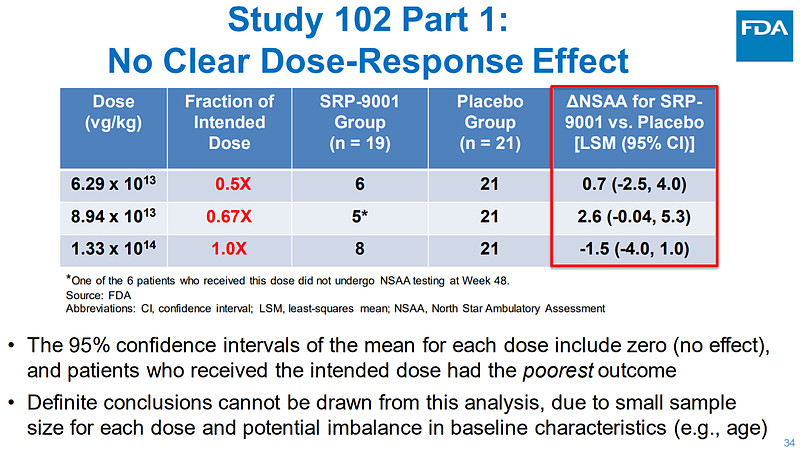
2)Sarepta在研究中引入外部对照组(包括Eli Lilly和Sarepta做过其他研究的安慰剂组、以及历史上其他研究的对照组),这些对照结果可能有倾向性。

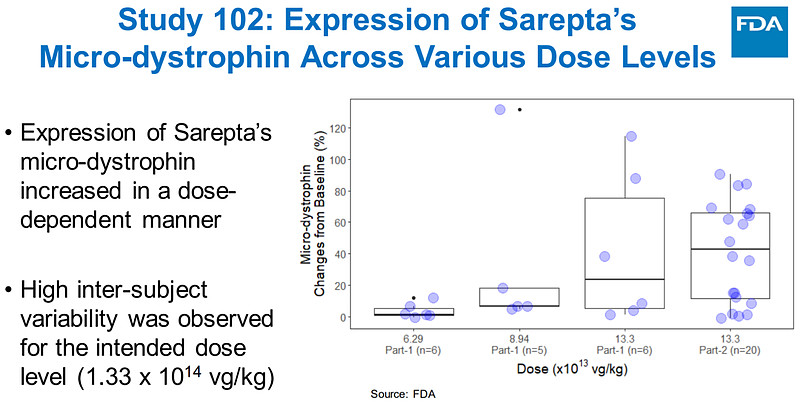
3)在Study 102中,micro-Dystrophin表达确实随着剂量而提升、但不同受试者之间的方差很大,最重要的是并未观察到NSAA变化与micro-Dystrophin表达量之间的相关性。
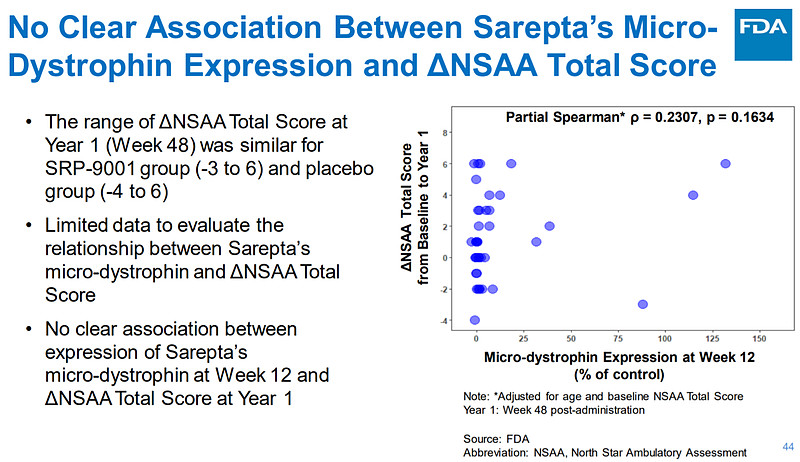
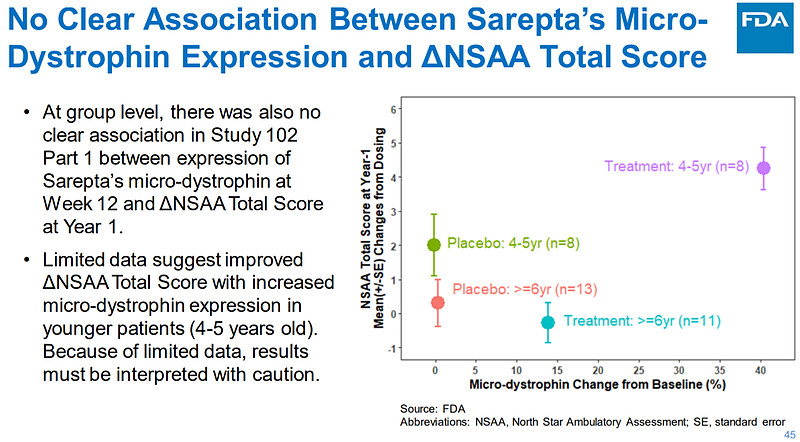
4)在开放标签试验中,NSAA变化优于随机对照试验,说明可能恰恰是开放标签设计影响了NSAA变化,而且在对基线影响进行调整后发现micro-Dystrophin表达只对NSAA变化起到11%的作用。


综合以上几点,FDA对于SRP-9001选择的替代终点有较多的顾虑。

3. 安全性
在SRP-9001的临床试验中,出现过的SAE包括肝毒性、心肌炎、免疫介导的炎症等,基本上也都是腺相关病毒载体基因治疗比较常见的不良反应。



4. 确证性临床
FDA对于确证性临床提出了一个非常贴心和前瞻性的问题:由于EMBARK研究需要观察52周,作为确证性临床的第一部分最后一例患者随访至少要到2023年9月,而届时通过加速审批的SRP-9001早已可以上市销售,考虑到DMD疾病的未满足临床需求和快速进展,这可能会导致一部分EMBARK研究的安慰剂组受试者不愿意继续留在临床试验中、转而直接寻求已上市的SRP-9001治疗,最终使得这个三期确证性临床的完成受到影响(或许这就是Sarepta前几个外显子跳跃DMD药物的确证性临床都杳无音信的原因?)

坦率说,当初SRP-9001上市前我也粗略看过FDA CTGTAC的这些会议材料,专家委员会的顾虑显然并没有阻碍加速审批,再次凸显了FDA在面对未满足临床需求时的渴望与灵活性。
然而在获得加速审批后不到半年,确证性临床的结果几乎是一一验证了当初的担心,不仅让这个品种是否能够获得完全批准变得扑朔迷离,而且让病毒载体、乃至整个基因治疗的前景再次蒙上阴影,甚至可能会让FDA对于加速审批和替代终点的脆弱信心再遭打击。
这款320万美元的天价药物,不仅是DMD患者的救命稻草,是否某种意义上也成为了基因治疗领域信心的晴雨表?