
撰文丨不器
编辑丨于靖
为期3天的听证会,14比1的投票结果——FDA产科、生殖和泌尿科药物咨询专家委员会这一压倒性的倾向,透露出早产预防药物Makena“命不久矣”的未来。
此前,FDA药品审评与研究中心(CDER)与Covis分别发布了简报文件,阐明各自立场。CDER用近百页的篇幅,详细解释其为何认为Makena应被撤市。与之相对,Covis坚定地为自家产品站台,还希望以各种方式给Makena“续命”。而最终的听证会现场,让双方的分歧更加凸显。
10月20日投票后的6个月,等待Makena的是FDA的最终裁决。某种程度上,这场听证会和结果走向,不仅仅代表着备受争议的Makena 10余年来的重要转折,背后更是作为全球标杆性的药品监管机构FDA如何“自我革命”。
01 有效性的争议,压倒性的投票
听证会讨论的核心问题之一是,确证性试验是否证实Makena对早产并发症新生儿发病率和死亡率的临床获益。
Makena最初在“试验002”(Meis试验)的基础上获得加速批准。这项研究从美国19家大学附属临床中心,纳入463名有自发性早产史的单胎妊娠女性,显示Makena活性成分己酸羟孕酮降低了复发性早产的风险。
但2020年12月,来自CDER的Silver Spring在NEJM发表文章,回顾了这次批准的不确定性。Spring称,早产预防药物只有在改善新生儿结局的情况下才有临床意义。可是,“试验002”中,Makena和安慰剂在有关新生儿结局的复合终点方面无显著差异,至于试验评价的15项新生儿并发症指标有3项发生显著改善,也存在不小假阳性的概率。
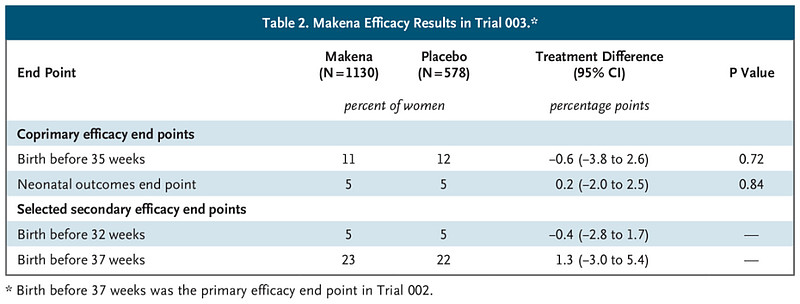
这就带出了Makena上市后的确证性试验,“试验003”(PROLONG试验)。跟前者不同,“试验003”特别纳入了新生儿结局终点,以此确认Makena对新生儿的临床益处。2018年,“试验003”得出结果,并未能证明Makena对早产的替代性终点、新生儿结局有影响。
Covis首席执行官Raghav Chari在此次听证会上回应说,FDA所引述的研究并不能简单对比,因为二者的入组人群比例不同。
支持加速Makena批准的“试验002”中,很多女性属于美国更容易早产的黑人群体,而该人群在确证性试验“试验003”显然小得多。Covis提交了一项后续试验表明,Makena有益于部分患者,包括黑人女性。
这正是最终投票环节,那唯一反对Makena撤市的意见立足点。Cassandra Henderson是一名纽约的母婴医学顾问,她认为,这种药物对某些人群确实有效,并重申了Covis的担忧,即黑人女性可能无法获得这种药物。因此,另一项研究进行之前应该让Makena继续留在市场上。
前述Spring发表的文章中,CDER承认“试验002”和“试验003”尽管试验采用相同的纳入标准,但两个研究人群的人口统计学特征有差异。
“试验002”仅在美国开展,而“试验003”中有75%的患者来自其他国家。由于“试验003”的规模较大,该试验纳入的美国女性人数(391人)与“试验002”的总人数(463人)相似。然而,CDER具体分析了“试验003”中美国和非美国参与者数据,均未发现Makena有效的证据,因此地理区域不能成为解释原因。
文章还进一步回应了关于黑人女性的占比问题。黑人女性在“试验002”中的比重为59%(273人),但在“试验003”中仅占7%(114人)。这114人有113人都来自美国,占美国全部参与者的29%。
这些差异确实值得注意,毕竟既往数据显示,美国黑人女性发生早产的风险较高。不过,CDER强调,其在“试验003”美国黑人女性或美国非黑人女性群体中,均未发现Makena有效的证据——这与Makena在该研究的非黑人群体所表现的无效趋势一致。因此,CDER表示,“试验003”中较低的黑人女性比例并不能解释Makena的有效性前后不一致。
除了黑人女性维度,CDER根据水平高低,特别统计对比两次研究中某些早产危险因素的参与者所显示的有效性,发现Makena在任何风险人群中均无疗效证据。
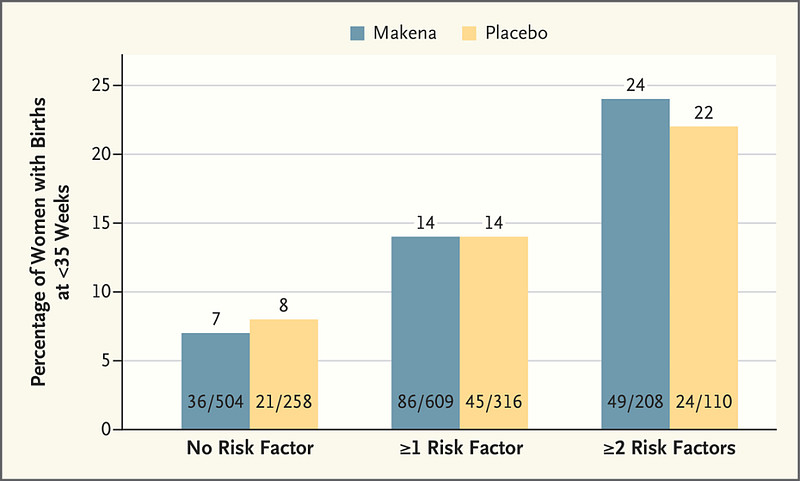
故而,FDA新药办公室主任Peter Stein的总结才指出,Covis在确证性试验的子集中所谓“重要发现”,“不是强有力的、可靠的观察结果,不是可靠的证据,也不是监管决策或指导临床实践决策的依据”。
针对Makena确证性试验对早产并发症新生儿发病率和死亡率获益问题上,15位专家都投了反对票。另一项核心讨论,即现有证据是否表明Makena对降低有单胎自发性早产史的单胎妊娠女性早产风险的批准适应症有效方面,13位专家持反对意见,剩余2位分别投了支持票、弃权。
02 临床亟需能否成为“挡箭牌”?
当然,专家委员会之外,支持将Makena保留在市场上的声音并不少见。
同样在NEJM,同样是2020年12月,来自妇产、公共卫生等领域的3位专家,Michael F. Greene、Mark A. Klebanoff、David Harrington,联名发表了FDA不应该撤回Makena的看法。这篇文章提议,需将Makena放在更广阔的背景下讨论。
上世纪90年代,美国缺乏经批准用于预防有早产史的女性再次早产的药物。于是,美国国家儿童健康与人类发展研究所(NICHD)赞助了将17α-羟基孕酮己酸酯(17-OHPC)这种合成孕激素作为应对方案的可能性。Makena便是出自这一研究。
“试验002”成果发布后,美国妇产科医师学会、美国出生缺陷基金会都强烈支持在适当的临床情况下使用Makena。这些建议,使得Makena单独在美国开展确证性试验的患者招募变得困难,因为卫生保健提供者和患者都认为缺乏平衡。最终,导致“试验003”的研究队列与原始研究不一致。
3位专家提醒,“试验003”来自9个国家的1708名参与者,黑人或非裔美国人中合格的新生儿结局终点事件总数为9例(Makena组69例中有6例,安慰剂组40例中有3例)。“这并不是一个可靠的数据库”,“根据如此少量的数据估计治疗效果可能会导致错误”,并且加速批准后Makena的“广泛使用并没有发现重要的安全信号”。
这番论述的潜台词是,一旦Makena被撤市,这些在“试验002”中表现出受益的群体将面临无药可用的境地。果如此,早产带来的身体和家庭打击可能进一步在不同人群中被放大。
“当大多数群体从某种药物中获益甚微,但面临严重医疗问题风险最大的少数群体似乎获得了显着利益时,任何最终导致无法获得该药物的决定都应谨慎进行。”联名文章总结说。
可此次听证会的专家组成员Susan Ellenberg不这么看:“未得到满足的需求不是将一个你不知道是否有效的药留在市场上的理由。”她不认同将Makena从市场上撤销就无法进行新的研究,相反,“如果在进行试验时让它继续留在市场上,反而可能会阻碍其它药物的开发”。
Stein也先将临床需求放在一边,他表示,“缺乏获益证据,且缺乏有效性证据,我们现在只剩下风险了”。
Chari呼吁,FDA将Makena的标签范围缩小到Covis在事后分析中确定的高风险患者,并在对这一高风险人群进行随机对照临床试验时将药物保留在市场上。按照计划,这一试验大约招募400人,需要4到6年完成,此外还有一项观察性研究,以建立胎龄和新生儿结局之间的关系。
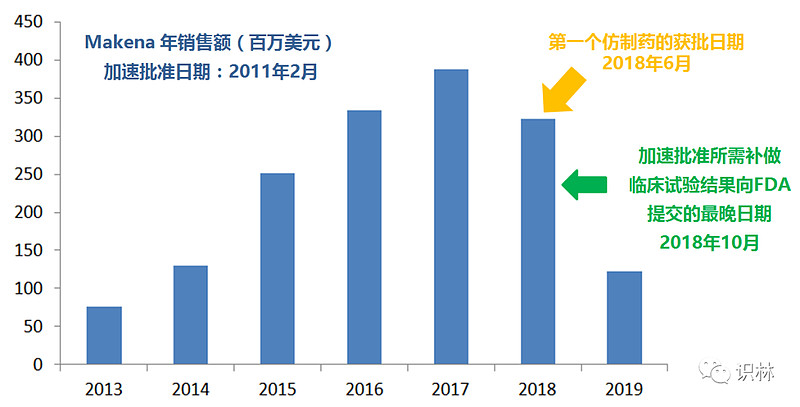
但这套说辞已经没有多少说服力了。自从2011年上市,Makena的销售额逐年增长,并在2017年达到峰值3.9亿美元。另一厢,“试验003”从2009年10月启动,直到2018年才完成,这可以说是“踩点”——FDA的加速批准信件,将Makena确证性试验的数据提交期限最迟定在2018年10月,而这只是提交,并非FDA决定撤销加速批准的日期。
“Covis声称可以在4到6年内完成另一项试验。我对此表示怀疑。对未来招募的最佳预测指标是过去的表现。在没有有效性证据且治疗药仍然可用的情况下,期待快速招募患者几乎是异想天开。”Stein反对说。
2018年之后,Makena仍在早产预防市场大行其道。根据BioPharma Dive的统计,Covis和该药物的原始所有者AMAG在Makena的销售中赚了大笔钱。2018年至2021年期间,仅来自医疗保险(Medicare)和医疗补助(Medicaid)的收入就达到了7亿美元。
想象一下——一款疗效和安全性都迟迟未能通过确证性试验的药物,却堂而皇之地上市销售10余年,赚取超10亿美元——就可以知道,此次听证会专家的压倒性撤市诉求不是空穴来风。
03 Makena事件:句号划在别处
如何说“放任”Makena在确证性试验上一拖再拖,倒还因为缺乏资料而情有可原;那么,“试验003”结果公布后,FDA直到2020年10月提议撤销相关加速批准,2022年11月才召开听证会,这个过程的曲折就难免不暴露出制度沉疴。
1992年,FDA设立加速批准(Accelerated Approval)程序,针对治疗严重疾病、相对于现有的疗法具有重要治疗优势的药物,这一通道提供了方便之门:不同于传统的终点,监管机构采用可预测临床益处(clinical benefit)的合理替代终点或中间临床终点(intermediate clinical endpoint),来作为审评依据。
Makena正是借助加速批准而提前上市的。彼时,FDA认为,复发性早产发生率降低是在合理范围内可能预测对新生儿有益的替代性终点。可事实证明,这种替代不一定能一一验证。对于携有安全性问题的药物来说,两者之间的偏差更加凸显。
今年4月,FDA召开的一次专家讨论会提到,部分PI3K抑制剂的随机试验中,ORR(客观缓解率)和PFS(无进展生存期)的数据证明相关药物可获益,总体生存期(OS)的结果却指示其具有潜在危害。
我们固然不应据此将加速批准制度“一棍子打死”,要求FDA审评专家做出完全准确的预判,但是,当事后证明先前决定有误,制度的自我纠偏,理当是可预期和实现的。结果呢?
根据NPR的调查,在最初的20年里,加速批准成为一种可选项,但FDA每年只通过这一途径批准少量药物。2012年的一项法律将FDA的政策固定下来,加速批准的数量激增。1992年只有一款药物获得加速批准,2020年这个数字增长到了49款。
作为附加条件,获得加速批准后,药企需要在限定时间里完成确证性试验,否则上市药物将被撤销。NPR分析,目前,有42%待完成的确证性试验(50项),要么在加速批准后花了一年多的时间才开始,要么根本没有启动。其中,19项有待完成的确证性试验,甚至在药物获得加速批准后的3年以上时间里仍未开始;有4项拖延超过了10年。
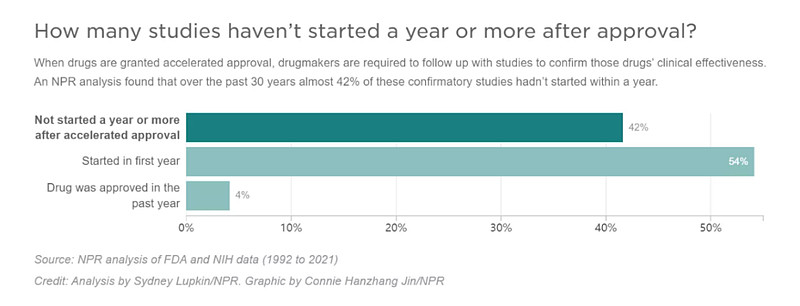
这意味着,许多在获得加速批准后上市的药物,有时会在患者、医生或监管机构不知道它们是否真的有效的情况下使用多年。
“下架”一款加速批准的药物并不简单。纵观先例,从咨询委员会投票撤销适应症,到FDA局长做出最终决定,基因泰克的Avastin乳腺癌适应症的撤销用了16个月。而从FDA提议撤销Makena算起,已经过去了两年。
接下来的问题是,Makena会否成为第二款FDA经过听证程序撤市的药物。答案或许不用等待太久。据Pink Sheet报道,FDA局长Robert Califf和该机构首席科学家Namandjé Bumpus将在6个月内,对Makena的撤市做出最终决定。
按照惯例,FDA不需要遵循咨询委员会的决定,但其中的不确定性也不会太大。随着Califf二度出任FDA局长一职,这个监管机构正大刀阔斧地推进改革,重拾公信力。再次就任前,Califf公开承诺,“FDA有责任确保这项工作(追责、撤市)不会随着最初的批准而结束”。
处罚不符合加速审批要求的药企,已被FDA提上日常,成为其今年计划指导文件的清单。值得注意的是,该机构的2023年预算文件包括立法提案,这或将给予FDA更多的权力来解决试验拖延的问题。
因此,与其将句号划在Makena的撤市决定上,不如多关注FDA对于加速批准等监管制度的重构。
比方说,“试验002”的设计无法确定Makena对新生儿是否有益,FDA本可以要求药企在药物审批前开展新生儿结局的确证性试验,却由于考虑了早产领域的临床亟需等因素而予以提前放行。后续,FDA有无收紧审批条件的可能?或者,FDA会不会选择性地确定有足够证据支持的临床替代终点?药企又该如何应对?……
上述问题,恐怕才是这款10余年“补票”未果的早产预防药物,给业界遗留下的更有价值的思考。
参考资料:
1.撤市倒计时:首款FDA加速批准早产预防药,与十年争论的遗产;同写意
2.“加速批准”迈向而立之年,FDA如何应对信任危机?;同写意
3.数据里的加速批准:FDA“放水”30年,验证性临床缘何难以启动?;同写意
4.FDA adcomm again votes to pull controversial pre-term birth drug from the market;Endpoints News
5.FDA opens case to withdraw controversial drug for preterm births;BioPharma Dive
6.Withdrawing Approval of Makena — A Proposal from the FDA Center for Drug Evaluation and Research;NEJM
7.Preterm Birth and 17OHP — Why the FDA Should Not Withdraw Approval;NEJM