应同写意邀请,泛生子首席医疗官胡云富博士在同写意主办的“第三届全球前沿技术大会”上做了《中美伴随诊断监管政策与注册申报策略》报告,本文系根据报告内容整理,并经报告人确认。

为什么要做伴随诊断?
伴随诊断(Companion Diagnostic,CDx)通过对分子标志物进行检测,为相关药物的安全、有效使用提供必不可少的信息。对医生和患者来说,通过伴随诊断可以为患者筛选出有效的治疗方案,降低不必要的毒副作用、节约治疗费用和时间;对于药企来说,通过伴随诊断筛选靶向药物的用药人群可以帮助提高临床成功率、缩短研发周期、降低研发成本。
通过一个比较简单的例子就可以看出伴随诊断在药物研发中的意义:当药效与对比药物相比差异较小时,通过伴随诊断精准筛选病人,可以把差异拉大,从而帮助药物提高临床成功率。另外,如果对应的分子标志物在患者中出现的频率非常低,也需要有伴随诊断(图1)。
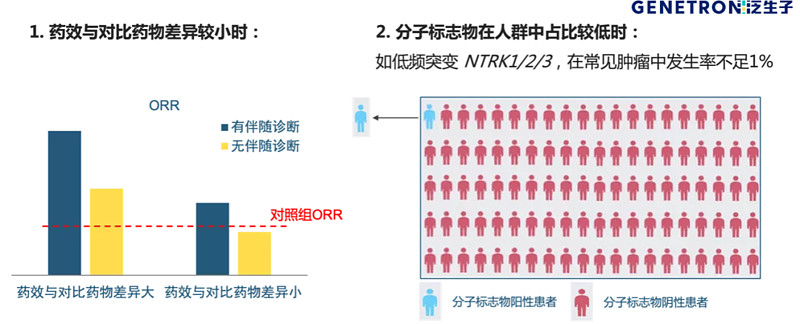
图1 特别需要做伴随诊断的情况
伴随诊断试剂开发的最重要的问题是其检测是否准确,尤其是低频突变的检测。如果一个伴随诊断有100%的灵敏度和99%的特异度,听起来似乎非常好,但99%的特异度就是说从100个阴性病人中会挑出一个假阳;对于1%突变频率的生物标志物来说,就是每测100个病人大约会挑出1个真阳和1个假阳入组,这样的话ORR一下就被假阳拉低一半;如果是98%的特异性,即每3个阳性结果里大约有1个真阳和2个假阳,ORR会继续下降。因此比较罕见的突变类型药物研发中,不仅要做伴随诊断,而且必须用非常准的伴随诊断。
伴随诊断的临床研究
美国FDA对伴随诊断临床研究有什么要求?从已获批的伴随诊断就可以体现出很多信息。今天我们重点介绍一下桥接试验的方法,因为现在很多新兴药物研发进展特别快、开发效果特别好,希望能够尽早上市——这时通常没有足够的时间做伴随诊断,伴随诊断的开发就需要做桥接实验。在FDA批准的所有伴随诊断预期用途中,有三十多个预期用途都是基于桥接试验批准的。
1、桥接试验
桥接试验非常简单,在药物临床入组时根据CTA(clinical trial assay)就可以在CTA阳性群体中收集到药效数据。这其中包括一部分CDx阳性病人与一部分CDx 阴性病人。对于做CDx的人来说,需要知道药物在用CDx筛选的人中药效如何,也就是CDx阳性结果。但其中有一组人群是CDx阳性CTA阴性,这部分人在药物临床中没有入组,推算这部分人群的药效是桥接实验的主要目的。
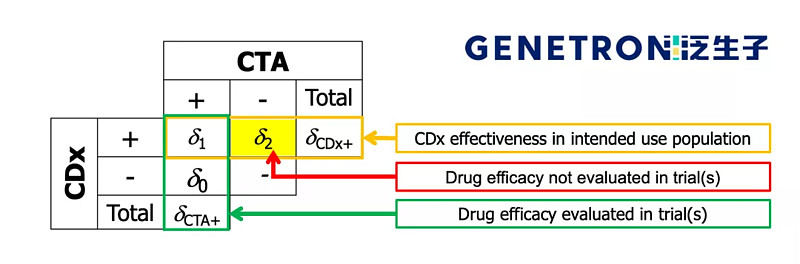
图2 桥接试验的方法
桥接试验的一大难点是合理地设置样本,包括样本的数量和来源。
最理想的情况是在药物临床入组时就注意保留样本,既包括阳性也包括未入组患者的阴性样本,并且阴性样本也要确保有知情同意书且允许重测。
在实际情况中,CTA阳性样本不一定全部可重测,此时如何满足美国FDA的要求?对于CTA阳性样本,FDA希望100%重测,但是在获批的CDx中很少是根据100%重测结果做桥接试验的。过去批准的伴随诊断桥接试验中,大约2/3的桥接试验阳性样本保留率在70%以上,还有约1/3的桥接试验阳性样本保留率在40%-70%之间。必须注意的是,对于丢失样本需证明其分布具有随机性,即在人口统计学和临床特征上与保留样本一致。为什么这样要求?举一个反例,假如保留的样本都是肿瘤大小比较大的,而丢失的样本都是肿瘤大小非常小的,因为前者比后者的检测更加容易,所以在这批样本上的研究结果不能代表未来在伴随药物预期使用人群里的效果。所以不仅要注意保留了哪些样本,还要注意丢失了哪些样本。
对于阴性样本,如果药物临床的阴性样本不可用,FDA允许采用外部研究补充阴性样本,但同样要注意补充样本能否代表伴随诊断预期使用人群。阴性样本的数量必须结合突变的发生率、检测的特异性等用统计方法分析确定。
2、同步开发
FDA强烈推荐伴随诊断与相应药物同步开发,在药物临床中采用申报的伴随诊断进行样本检测。同步开发是伴随诊断开发最理想的方式,可以保证同步开发的药物与伴随诊断试剂同时上市。如果伴随诊断的性能验证数据不全,美国FDA有PAS(获批后试验)机制来增补。
不管是桥接试验还是同步开发,在伴随诊断滞后无法与药物同时获批的情况下,FDA才采用PMC(上市后承诺)和PMR(上市后要求)的方式,要求药厂向FDA保证在药物上市之后在一定期限内满足完成伴随诊断试剂开发的承诺和要求。
3、后续研究(follow-on)
会场在座的可能有检测公司的同行,所以这里也简单介绍一下伴随诊断的后续研究开发途径。后续研究中,哪些产品可以作为参考方式用来对比?中国NMPA和美国FDA都要求只能与原研伴随诊断做对比,通过后续研究获批的伴随诊断不能作为其它伴随诊断的参考方法。判断一个伴随诊断是不是原研,只要看它获批时有没有临床药效数据即可。
研究方法有两种,一种是过去常采用的方法对比,比较简单。其中非常关键的是对差异的统计分析。可能很多人认为差异为零肯定可以满足要求,但实际上不太可能做到,因为从统计上来讲这意味着申报的方法优于参考方法,在这个实验里是不可能证明这一点的。什么样的差异是可以接受的?不论中国还是美国这点都需要和监管机构提前沟通确定。
另一种是近几年应用比较多的方法,叫非劣效性研究(Non-Inferiority Study)。非劣效性研究用参考方法对样本进行2次检测,用申报的伴随诊断对样本再进行1次检测,最后统计分析参考方法自身的一致性和申报方法与参考方法的一致性。方法内和方法间的一致性差异有多大可以容忍,也是需要提前和NMPA或FDA沟通确认的。
伴随诊断的临床合规
关于在美国开展伴随诊断临床试验的合规性要求,大家常有的一个疑问是什么情况下必须提交IDE,在这里也简单介绍一下。与药物临床中的IND类似,在美国开展的临床试验如使用未获批的医疗器械,都需要遵守IDE法规要求。但并非所有药物临床试验都需要向FDA提交IDE。是否需要向FDA提交IDE取决于药物的临床研究(包括依据分子标志物入组病人的抗肿瘤新药的临床研究)属于“NSR(无重大风险研究)”还是“SR(有重大风险研究)”,仅后者需要向FDA提交IDE。有无重大风险由IRB和FDA判定,一个重要的判定依据是:患者如果依据检测结果入组,是否将放弃其他已获批准或被认为有效的疗法。例如针对已经有药物上市的ROS1靶点,如果做一线新药临床,就是“有重大风险”;而针对没有有效疗法的耐药或难治性肿瘤新药临床,则可能被判定为“无重大风险”。
需要注意的是,“无重大风险”的临床研究仍然需要收集IDE要求的相关信息,包括用来入组检测方法的性能验证, 只是无需向FDA提交。FDA有权要求查看研究者的相关记录。
伴随诊断注册中美双报策略
临床试验完成之后就是中美双报的问题。中国加入ICH后,境外数据的可利用为药物审批提供了很多便利,但在分子检测方面有一点需要大家注意:不能向美国或其他任何国家提供任何在中国药物临床试验中收集的任何中国人的样本或遗传信息。因此,针对中美伴随诊断双报最简单、理想的路径,就是选择具有中美双中心实验室的诊断公司作为合作伙伴,使用同一个试剂盒,进行中美同步试验,并行两边的注册申报,该情况下可将国外数据合并到国内分析。而对于采用两个不同试剂盒、需要以桥接试验进行伴随诊断申报的项目,依然需要通过具备中美双报能力的合作者,完成两个试剂盒在临床试验及疗效数据上的比较,并分别在两地完成申报,该情况下必须将国外剩余样本合并到国内检测。
当前CDx开发过程也面临一些挑战,需要进行一些调整。留下几个问题,大家有兴趣的话可以共同讨论:如果使用多个LDT进行注册,如何递交IDE?桥接到哪些LDT?如何选择LDT阴性样品?如何使用已获批LDT/CDx进行其它伴随诊断比对试验?如何扩展已获批CDx的适应症?