6月17日,BioNTech在一份SEC文件中披露,从合作伙伴宜联生物获悉,美国FDA已部分暂停在研HER3 ADC药物BNT326/YL202的多中心、开放标签、首次人体I期临床(NCT05653752),该试验旨在评估BNT326/YL202作为EGFR突变晚期或转移性非小细胞肺癌(NSCLC)或HR+/HER2-乳腺癌(BC)患者的后线疗法的安全性。部分暂停影响了该试验在美国的新患者入组。

FDA向宜联生物表达了对BNT326/YL202的担忧,即在较高剂量下,BNT326/YL202可能会使受试者面临不合理的重大疾病或伤害风险。
为了满足FDA的要求,需要采取一些措施,包括审查临床和安全性数据、与该机构共享现有药理数据,以及在研究者手册中提供更多有关安全性发现的信息,包括在YL202-INT-101-01和YL202-CN-201-01研究中观察到的5级不良事件,也就是死亡事件。宜联生物已采取行动暂停在美国招募新患者,并满足FDA的要求。
YL202为靶向HER3的ADC,基于宜联TMALIN平台所设计。2023年10月,宜联生物就该产品与BioNTech达成海外合作授权,BioNTech向宜联生物支付7000万美元首付款及潜在超10亿美元总付款,获得YL202海外开发、制造和商业化的独家权利。
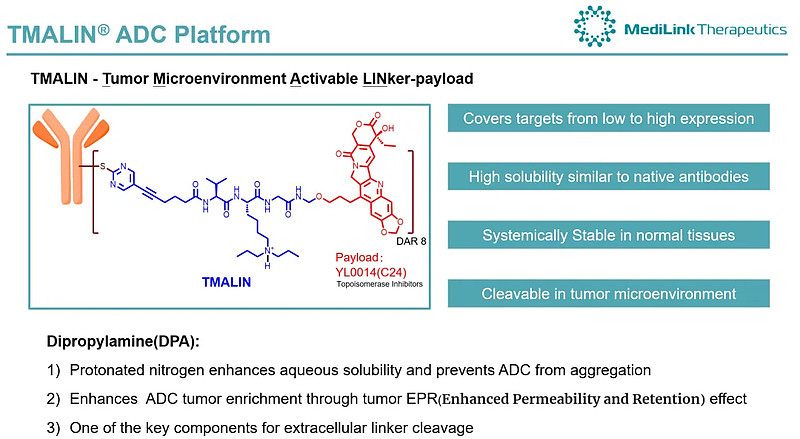
在今年ASCO大会上,宜联生物以壁报形式报告了YL202的临床数据。截至2024年4月16日,该项中、美同步开展的I期临床爬坡共计入组55人(非小细胞肺癌40人,乳腺癌15人),累计爬坡7个剂量组(0.5 – 5.5 mg/kg Q3W)。
疗效方面,全部剂量下共51例可评估病例,客观缓解率(ORR)为42.3%,疾病控制率(DCR)为94.2%。其中3.0mg/kg剂量组ORR为60.0%,DCR为100%。在EGFRm NSCLC末线人群中,全部爬坡剂量下ORR为39.4%(15/38);在HR+/HER2- BC末线人群中,全部爬坡剂量下ORR为53.8%(7/13)。
安全性方面,常见治疗相关安全事件包括白细胞计数减少(40%),中性粒细胞减少(31%)等血液学副作用。在爬坡过程中,5.5mg/kg剂量组下出现一例剂量限制性毒性(DLT)事件(三级粒缺伴发热)。在回填过程中,4.0mg/kg和5.5mg/kg剂量组下分别出现2例(中性粒和血细胞减少)和1例受试者死亡(新冠感染后间质性肺炎)。
近期热门研究报告下载
2024 ASCO 新药开发机会分析网页链接
ASCO ESMO 2024 信达生物 - 肿瘤管线临床数据更新网页链接
2024Q1 年度中国医院药品市场分析报告网页链接
GSK 2024年肿瘤领域介绍网页链接
更多相关报告和医药资讯持续更新,请持续关注医药魔方ByDrug 网页链接