原创 💧transparent
▉ 摘要
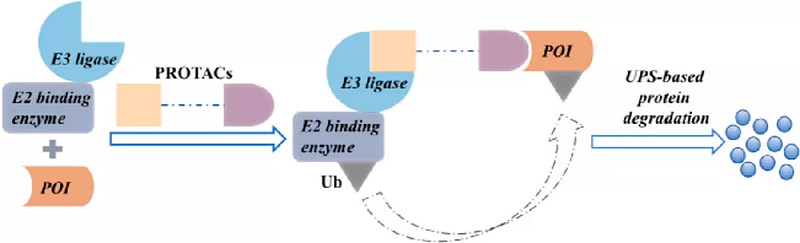
图1. PROTACs作用机理
在Crews等人开发出第一个蛋白降解靶向嵌合体(Proteolysis targeting chimera,PROTAC)分子后,PROTAC技术倍受好评1。最近,越来越多的PROTAC被用于新药的发现与开发。PROTAC体系通过一个灵活的接头偶联一个靶蛋白(POI)配体和一个E3连接酶配体。PROTAC与目标蛋白结合并募集E3连接酶,通过泛素-蛋白酶体途径降解整个靶蛋白(图1)2。

今天为大家讲解一篇出自昆明理工大学医学院的综述。该篇主要从药物化学的角度总结了最前沿的靶向降解蛋白激酶的PROTAC分子。
▉ 1. 靶向BCR-ABL
BCR-ABL已成为慢性粒细胞白血病(chronic myelocytic leukemia,CML) 中的潜在抗癌药物靶点。伊马替尼是第一个靶向 BCR-ABL的酪氨酸激酶抑制剂(Tyrosine Kinase Inhibitors,TKI)。随后,多种ABL激酶抑制剂,包括波纳替尼、达沙替尼、尼罗替尼和波舒替尼已被批准用于临床。尽管TKI的使用改善了CML的治疗结果,但长期治疗和耐药性仍是重大挑战4。
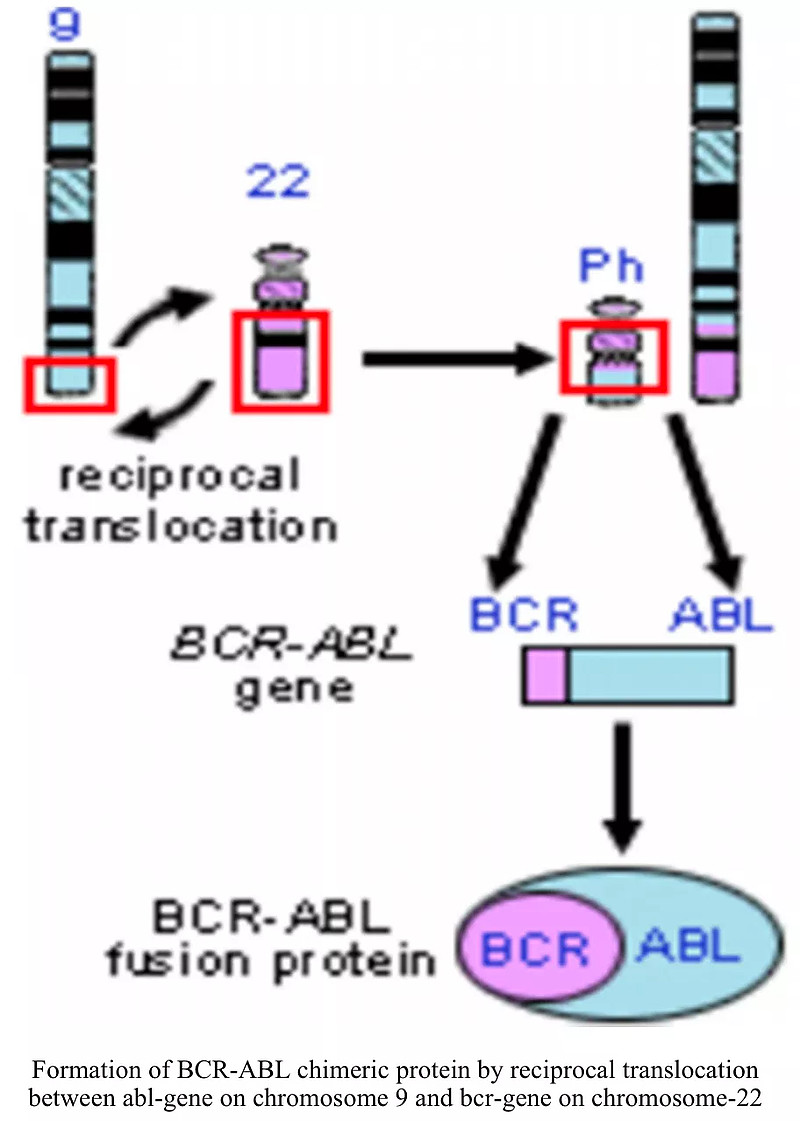
BCR-ABL融合蛋白形成过程
DOI:10.11648/j.crj.20180601.15
研究发现BCR-ABL的降解可能对CML治疗有益。Shimokawa等人5报道了一种新型抗癌剂SNIPER(ABL)-062以降解致癌蛋白(1,图2)。该化合物与cIAP1/2、ABL1 和 XIAP 具有很强的结合亲和力,导致BCR-ABL完全降解并抑制BCR-ABL介导的信号通路。
Zhao等人6推测优化接头结构可促进BCR-ABL和E3连接酶之间相互作用。基于这个假设,他们开发了一种强大的BCR-ABL降解剂SIAIS178 (2,图2)。SIAIS178在体外表现出明显的选择性并显着抑制BCR-ABL+白血病细胞的生长,在体内,对K562异种移植肿瘤具有显著抑制作用。
Yang等人7报道了针对BCR-ABL蛋白中达沙替尼、波纳替尼和asciminib三个结合位点的P19P (3,图2)。此外,Crews等人8发现GMB-475 (4,图2)能诱导蛋白酶体快速降解并表现出更高的敏感性
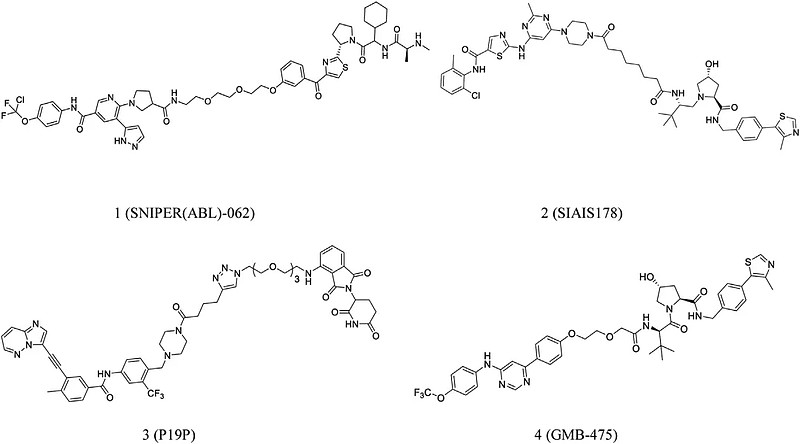
图2. 靶向ABL-BCR的PROTACs化学结构
▉ 2. 靶向PI3K-Akt
磷脂酰肌醇3激酶(Phosphatidylinositol 3-Kinase,PI3K)是细胞内脂质激酶,在细胞存活、增殖、增长、分化和迁移中发挥着作用,是潜在的抗癌靶点。PI3K分为三类,其中I类被广泛研究。大多数癌细胞表现出I型PI3K的过度激活突变,这些突变驱动肿瘤细胞增殖和存活9。
通过使用不同的接头将泊马度胺和哌嗪衍生物结合,Li等人10开发了新型PI3K-PROTAC。这些化合物可以显着抑制PI3Ka且IC50值在纳摩尔水平。其中最有效为化合物B(5,图3),它对PI3Ka的IC50为18 nM。此外,Tovell等人11报道了SGK3-PROTAC1(6,图3)用于选择性地降解SGK3。亚微摩尔浓度下的SGK3-PROTAC1即可在不同癌细胞系中诱导蛋白酶体介导的降解。
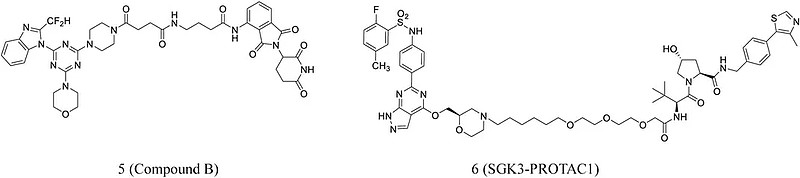
Figure 3. Chemical structures of PROTACs targeting PI3K/Akt. (Frontiers in Chemistry,2021)
▉3. 靶向布鲁顿酪氨酸激酶
布鲁顿酪氨酸激酶(BTK) 促进B细胞生长、成熟、迁移和凋亡。癌症、自身免疫或炎症可能是由BTK通路失调引起的12。目前为止,BTKs的研究取得了长足的进步,BTK抑制剂如ibrutinib,阿卡拉替尼和zanubrutinib已被FDA批准使用。然而,BTK抑制剂仍具有一定的局限性,如耐药性和脱靶效应。
最近,Buhimschi等人报道了MT802 (7,图4),它通过PEG接头连接BTK特异性配体和CRBN配体。与依鲁替尼相比,该化合物能更有效降解BTK。Jaime-Figueroa等人进一步修饰了CRBN配体结构并合成了SJF620 (8,图4),在小鼠中表现出优于MT802的药代动力学特征。Pan,Huang,Liu,Sun等人基于之前的工作,对化合物进行进一步优化,分别合成了PROTAC 7 (9,图4)、TL12-186(10,图4)、SPB5208 (11,图4)、P13l (12,图4)。
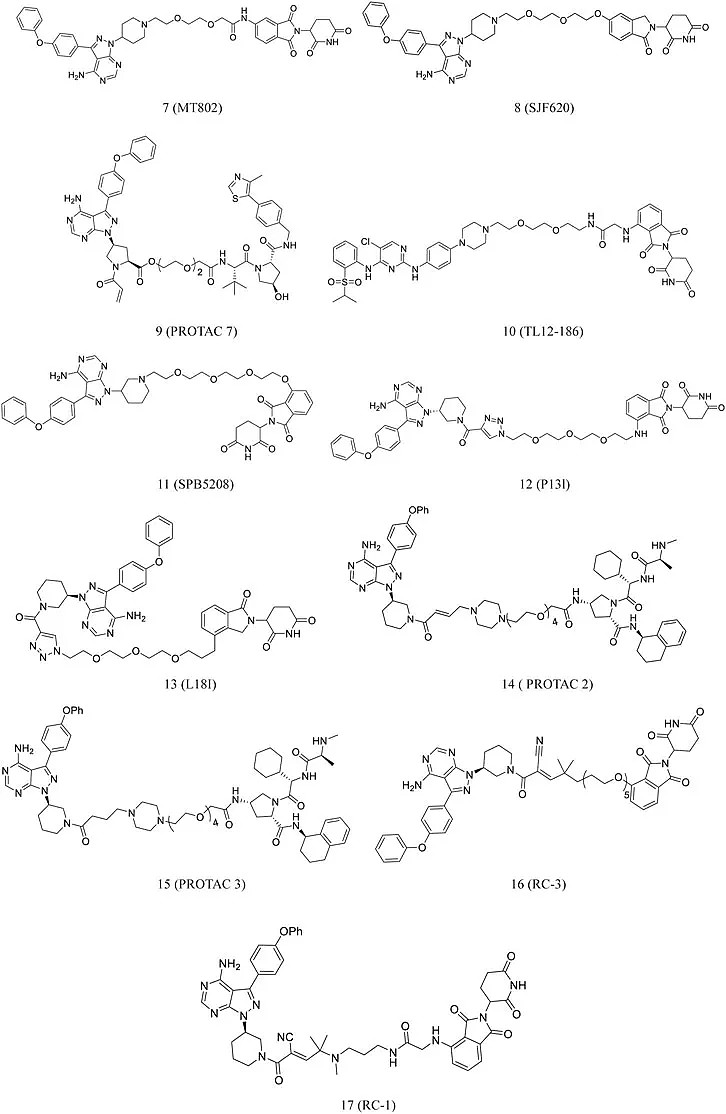
图4 靶向BTK的PROTAC化学结构
Sun等人还开发了L18I (13,图4),在30 nM时可诱导HBL-1细胞中耐依鲁替尼的BTK降解。令人兴奋的是,L18I在耐药肿瘤细培养的小鼠中发挥了明显的抗肿瘤作用。迄今为止,大多数报道的PROTAC是通过共价或非共价结合起作用的。不可逆共价抑制剂具有很强的靶标亲和力和高靶标占有率,并已在临床环境中取得成功。
然而,不可逆的结合可能会通过降低PROTAC的催化活性而降低抗肿瘤效力。Dittus等人选择共价抑制剂依鲁替尼和可逆结合类似物开发了PROTAC 2(14,图4) 和PROTAC 3 (15,图4)并对其BTK降解能力进行了调查。PROTAC 2不降解共价结合的靶标蛋白,而PROTAC 3降解其靶标蛋白。这一发现强调了催化对PROTAC介导的成功降解具有重要意义。此外,可逆共价抑制剂具有选择性且能增加抗肿瘤效力。为了提高PROTAC的功效,Gabizon和Guo等人分别合成了高效的、可逆共价的RC-3 (16,图4)和RC-1 (17,图4)。
▉4.靶向酪氨酸激酶受体
酪氨酸激酶(RTK) 在控制基本细胞过程和调节细胞间通讯过程的信号通路中发挥关键作用。EGFR作为一种RTK,与细胞凋亡、增殖、代谢和存活的调控密切相关13。EGFR的过表达和激活突变与几种癌症类型有关。EGFR的失控激活导致异常细胞生长,这可能会导致EGFR信号的内吞作用和细胞内的末端转运。最近,一些小分子和抗体靶向EGFR的治疗药物被开发用于癌症治疗14。迄今为止,FDA已向非小细胞肺癌患者提供了十多种EGFR抑制剂,但EGFR突变变体可能会影响药物疗效。
PROTACs能降解几种RTK,包括 cMet、HER2、EGFR以及EGFR和c-Met的多个突变体。Konecny通过将EGFR结合元件拉帕替尼与E3连接酶结合配体缀合合成 PROTAC 1(18,图5)。在较低纳摩尔浓度下,该化合物就表现出细胞膜穿透性和EGFR降解作用。Zhang等人开发了含有吡啶并[3,4-d]的新系列EGFR降解剂。在HCC827细胞中,PROTACs 2(19,图5)和 10(20,图5) 有效降解EGFR并显着诱导HCC827细胞凋亡。Zhao、Zhang、He分别基于EGFR靶点设计合成了特异性的PROTAC降解剂:P3 (21,图5)、14o (22,图5)和16c (23,图5)。Cheng等人基于吉非替尼化学结构,开发了两个分别由VHL和CRBN招募的EGFR降解剂MS39 (24, 图5) 和MS154 (25,图5),两种化合物都能选择性地诱导突变型EGFR的降解。
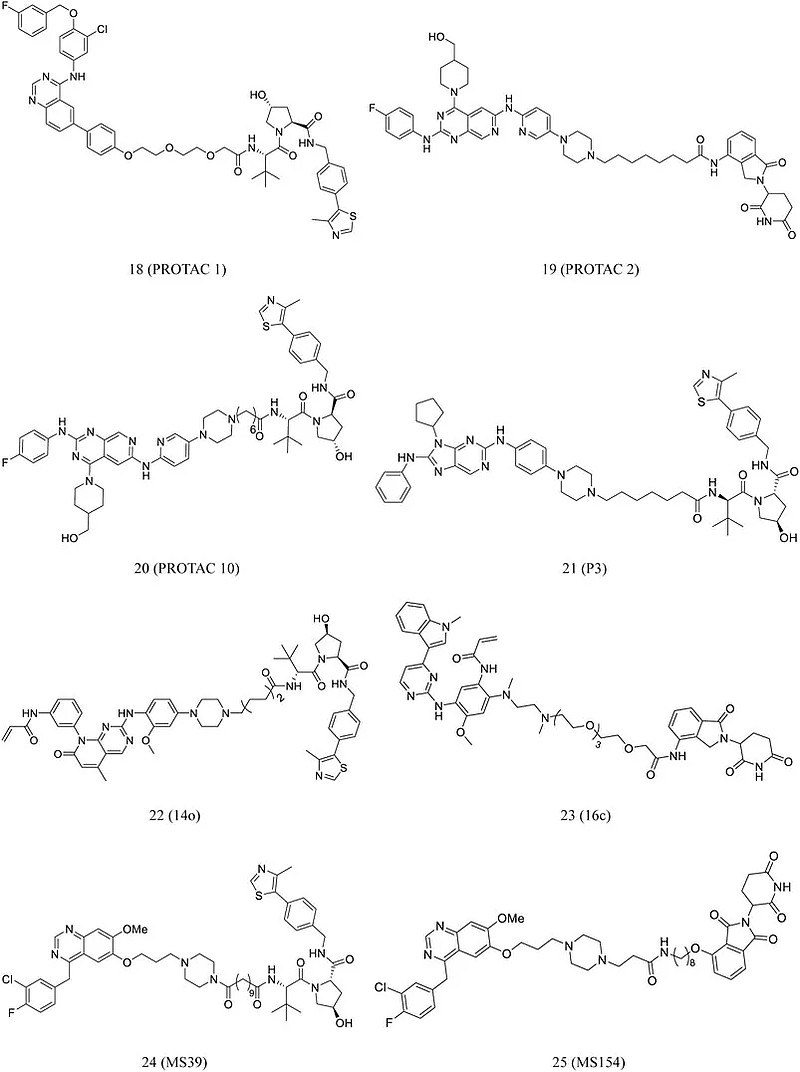
图5 靶向RTK的PROTAC化学结构
▉5. 靶向粘着斑激酶
粘着斑激酶 (FAK),一种胞质酪氨酸激酶,于1992年首次报道15。FAK通过激酶依赖性方式促进肿瘤生长、侵袭和转移。有研究表明原发性和转移性癌组织中FAK表达及活性的增加不利于患者生存,抑制FAK可以潜在地阻止肿瘤进展16。
目前,小分子FAK抑制剂的临床效果已进行研究调查。不幸的是,FAK具有激酶依赖性和激酶非依赖性功能,这意味着FAK抑制剂可以拮抗其激酶依赖性功能,但不能拮抗激酶非依赖性功能。然而,靶向FAK的PROTAC可以同时抑制激酶依赖性和激酶非依赖性功能17。
目前,PROTAC-3(26,图6) 最有前途的降解剂,在抑制人三阴性乳腺癌细胞中的FAK信号传导和细胞迁移和侵袭方面优于临床FAK抑制剂defactinib。Popow等人开发了针对PTK2降解的高选择性的PROTAC。通过聚乙二醇接头将高选择性的PTK2抑制剂与CRBN或VHL配体进行共轭连接,从而分别开发出选择性的PTK2 降解剂BI-0319(27,图6)和BI3663(28,图6),两种化合物都可以选择性地降解PTK2 蛋白。此外,基于FAK抑制剂和CRBN E3配体,Gao 等人开发了一种新型的FAK降解剂 FC-11(29,图6),其在各种细胞系中孵育8小时后即可完全降解FAK蛋白。
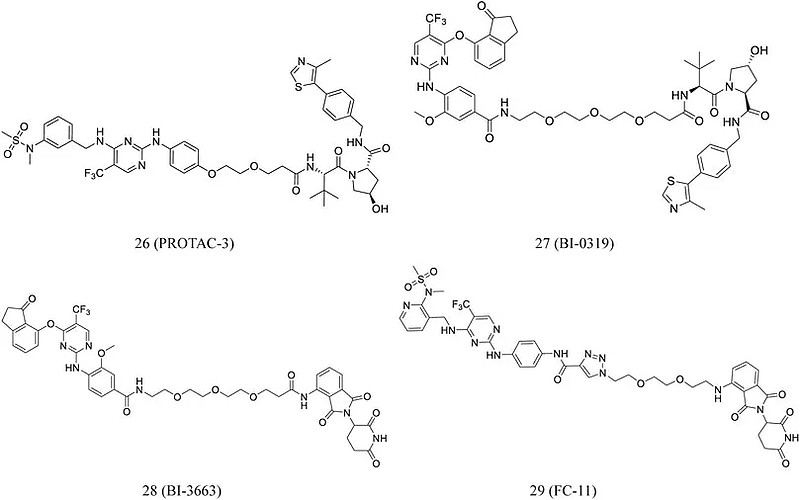
图6 靶向FAK的PROTAC化学结构
▉ 6. 靶向细胞周期蛋白依赖性激酶
有研究报道,在细胞周期蛋白依赖性激酶(CDK) 家族中,CDK1-4、CDK11和CDK6调节细胞周期,而CDK7-9主要调节转录18。在早期的研究一直在探究小分子CDK抑制剂对各种癌症的影响,例如急性髓性白血病 (AML)、BC、NSCLC 和前列腺癌(PC)19。在肿瘤模型中,小分子CDK抑制能抑制细胞生长并促进细胞周期停滞,从而促进细胞凋亡,是一种有前途的癌症治疗策略。
2015年,第一个选择性CDK4/6抑制剂palbociclib被FDA批准用于治疗转移性BC。此外,终末分化的细胞表现出增加的 CDK9 表达。一些研究发现,选择性 CDK9 抑制剂具有治疗包括癌症的多种疾病的潜力。大量证据表明CDK9是一种潜在的肿瘤治疗靶点。既往研究表明,不良反应和毒性限制了CDK9抑制剂在临床上的广泛应用。因此,有必要制定可行的治疗策略20。
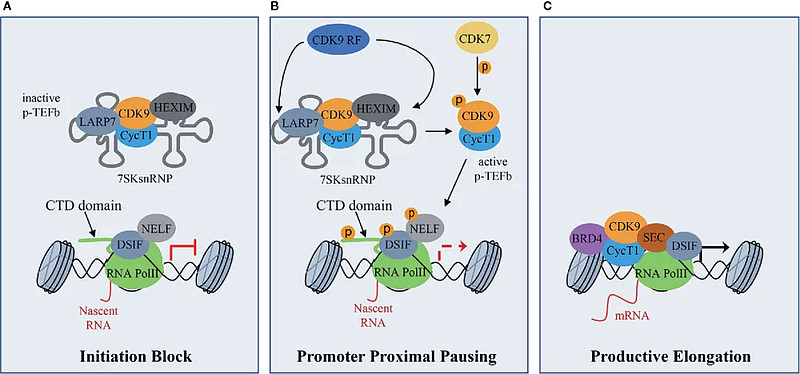
CDK9在RNA聚合酶II的激活和基因转录方面的功能
目前,已有多种选择性CDK9抑制剂被开发出来,如PROTAC 3(30,图7),B03 (31,图7),PROTAC 11c (32,图7),THAL-SNS-032 (33,图7)。Zhou等人构建了一种双重降解剂F3 (34,图7),其可以同时降解CDK9和CDK2,通过有效阻断PC-3细胞的S期和G2/M期来抑制细胞增殖。此外,它还降低了各种癌细胞系中CDK2/9的活性,表明其具有很好的抗癌潜力。
CDK8调节基因转录,因此在干细胞功能、炎症和免疫反应中发挥重要作用。Hatcher等人依据高选择性CDK8抑制剂JH-VIII-49的结构,进一步开发了一种二价降解剂JH-XI-10-02 (35, 图7),它通过募集 Jurkat细胞中的E3连接酶可诱导CDK8的显着降解。同理,CDK4/6和CDK2也是癌症治疗中的靶点,基于此,Zhao和Wei等人分别设计开发了CDK4/6抑制剂pal-pom (36,图7)和CDK 2/4/6抑制剂PROTAC 3(37,图7)。Wei等人在PROTAC 3的结构基础上修改得到了具有良好的口服生物利用度的prodrug 11 (38,图7)。接着,Brand等人报道了一种基于邻苯二甲酰亚胺的降解剂BSJ-03-123 (39,图7),这种降解剂可以实现对CDK6蛋白质组的选择性降解。
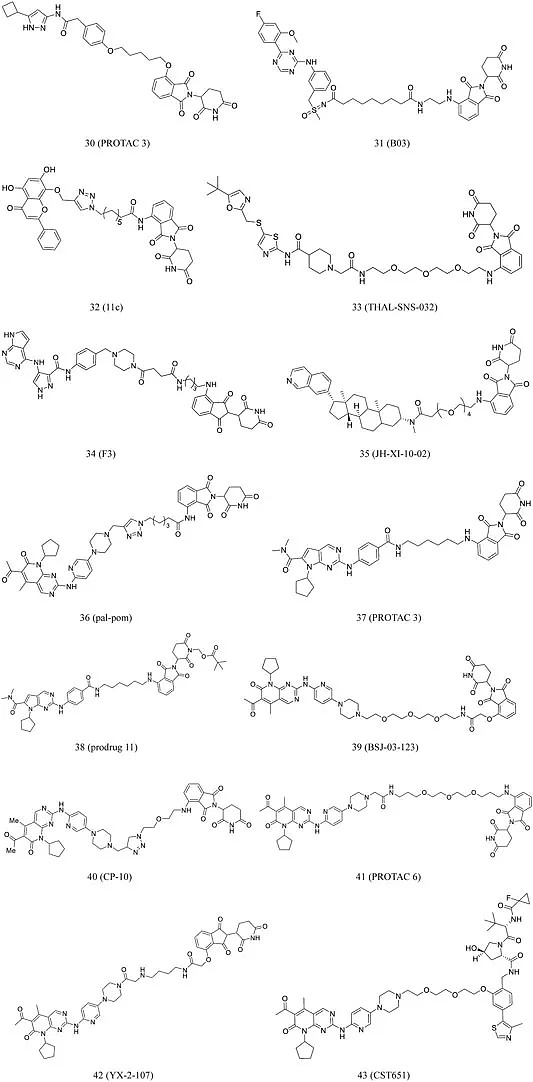
图7 靶向CDK的PROTAC分子化学结构
此外,为了了解 PROTAC 与靶标之间的结合机制,研究者们通过评估结合亲和力、空间方向和接头长度,得出最强大的PROTAC为CP-10(40,图7) ,它同时阻止了几种造血癌细胞的增殖。此外,据报道,这种化合物可以从突变和过表达中降解CDK6。通过使用不同长度和组成的柔性接头将 palbociclib 与泊马度胺缀合,Rana等人合成了PROTAC 6 (41,图7),它能选择性降解CDK6且不影响其他CDK。另外,YX-2-107 (42,图7)和CST651(43,图7)也可以选择性诱导CDK6降解,并在几种人和鼠癌细胞中均已证明有效。
▉ 7. 靶向MEK1/2
先前的研究表明,RAS-RAF-MEK-ERK参与了多种细胞过程,如增殖、分化、凋亡、迁移和代谢21。此外,MEK1和MEK2在调节ERK的活性中起重要作用。MEK1或MEK2受体突变诱导的 ERK信号过度激活与人类癌症有关。迄今为止,多种靶向MEK1/2的药物如司美替尼、考比替尼、曲美替尼,已被FDA批准用于治疗癌症。然而,患者对MEK1/2抑制剂会逐渐产生耐药,届时需要开发新疗法。
Wei等人结合MEK1/2抑制剂PD0325901开发了MS432 (44, 图8),该化合物能持久地有效降解MEK1和MEK2且具有浓度和时间依赖性。在体外,其能够有效阻止ERK的磷酸化。Vollmer等人根据PEG接头和MEK抑制剂结构设计合成PROTAC 4 (45,图8),虽然 PROTAC 4在抑制ERK1/2 磷酸化方面效果较差,但在抑制A375细胞增殖方面更有效。
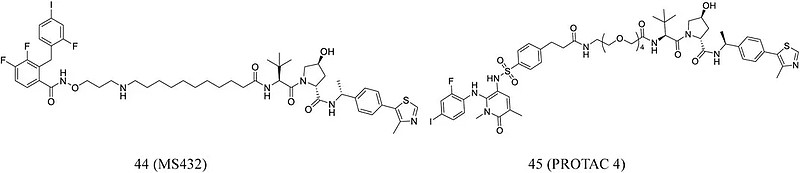
图8 靶向MEK的PROTAC分子化学结构
▉ 结论
PROTACs可以破坏靶蛋白的酶促和非酶促功能,是一种解决获得性耐药的新方法。近年来,ROTAC技术取得了重大进展,这种策略特别适用于蛋白激酶。蛋白激酶的异常功能与癌症密切相关,因而这些激酶作为抗癌药物开发治疗靶点具有重要意义。
然而,突变激酶和耐药性的出现仍然是限制激酶抑制剂功效的主要因素。在过去的几十年中,许多选择性靶向激酶的配体作为竞争性或变构抑制剂被开发出来,这为PROTAC分子的设计打下了基础。此外,PROTACs介导的激酶降解也可用于调节磷酸化。然而,它们仍然存在一定的局限性,需要对其作用机制进行进一步研究,以更好地开发下一代PROTAC。
参考文献:
《Targeting Protein Kinases Degradation by PROTACs》