核心观点
创新药出海的内在逻辑:医疗支出差距、药品价格差距、药品峰值差距和快速回笼资金
• 创新药出海的内在逻辑包括以下三点:第一,中美医疗支付能力差距显著,海外市场空间巨大;第二,创新药在国际市场上的售价通常更高,中美药品销售药品峰值存在巨大差异;第三,首付款快速回笼资金,解决企业燃眉之急,并加速推进海外临床试验。
创新药出海的三要素:市场空间大,疗效和安全性优势、先发优势
• 市场空间巨大,可有效解决未满足的临床需求。市场空间的潜力是MNC最在意的问题,如强生携手传奇生物共同推进西达基奥仑赛的商业化,强生预计西达基奥仑赛全球销售额有望达50亿美元。
• 临床有效性及安全性较目前标准疗法优势明显。药物的临床疗效和安全性优势是药品拓展市场的关键,最终反映出医生和患者对药品的信赖度。
• 临床进展居前,先发优势明显。药品的先发优势对于药物的放量至关重要。根据《2020年度中国抗肿瘤新药临床研究评述》,全球范围内同一靶点药物,首个上市产品可以获得45%的市场份额,第二至第四个上市的产品分别可以获得27.9%、14%以及11.3%的市场份额。
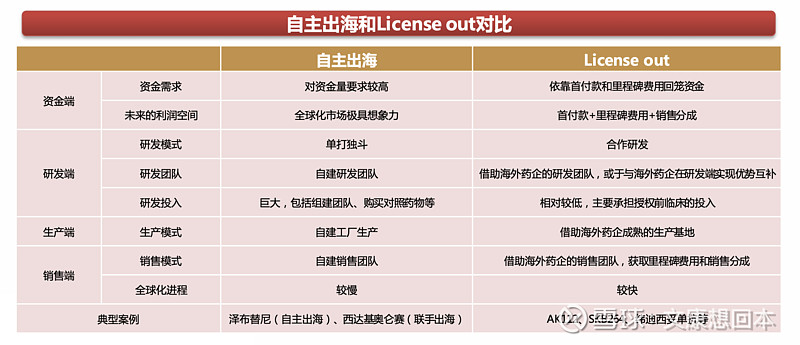
出海可分为自主出海和License out,目前,License out是出海的主要模式
• 自主出海即中国药企凭借自身的团队在海外国家和地区开展临床试验,申报上市,获批后展开销售。License out则是中国药企将自身产品的海外权益或全球权益许可给以欧美跨国药企为代表的制药企业,获得首付款和里程碑费用。海外药企接过接力棒后,负责海外市场的临床开发、申报上市、生产及销售工作。
自主出海难度较大,但商业化前景广阔,百济神州和传奇生物已经示范
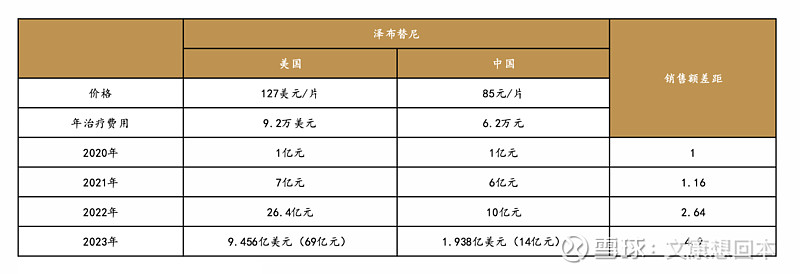
• 百济神州:泽布替尼是百济神州旗下一款BTK抑制剂,于2019年在美国上市,是国产创新药自主出海的标杆。2023年,泽布替尼美国收入69亿元,中国收入14亿元,差距达4.9倍,销售额差距的加大反映出中美市场的巨大差异,一方面是中美药品治疗费用的差异,另一方面则是药品峰值的差异。
• 传奇生物:2022年2月底,西达基奥仑赛通过FDA批准上市,成为首款获得美国FDA批准的中国CAR-T细胞疗法。2017年12月,强生子公司Janssen与传奇生物签订了独家全球许可和合作协议,以开发和商业化西达基奥仑赛。2023年,西达基奥仑赛全球销售额达4.98亿美元。
License out:金额不断提升,数量持续攀升
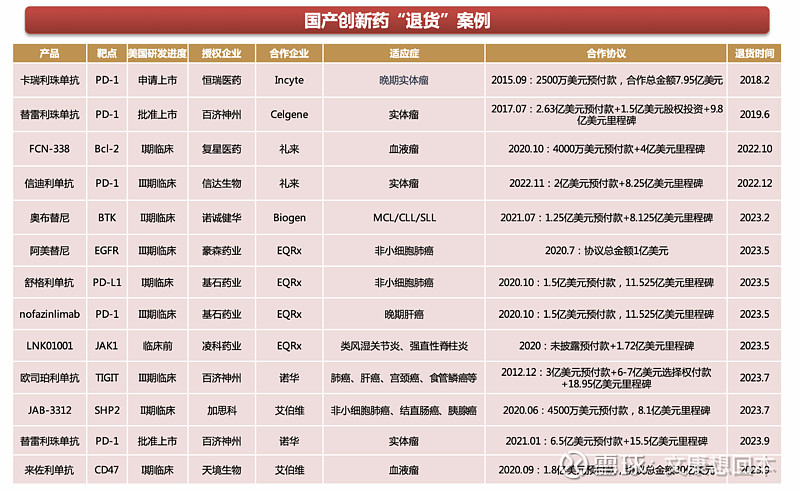
• 随着国内新药创制水平的不断提升,国产创新药的国际认可度稳步上升,国产创新药License out金额持续攀升。超过30个License out项目总交易金额超5亿美元,其中超20亿美元的项目包括科伦药业的七个ADC项目(94.7亿美元)、百利天恒的BL-B01D1(84亿美元)、康方生物AK112(50亿美元)、荣昌生物的纬迪西妥单抗(26亿美元)等,国产新药License out交易金额正快速提升。
• License out下的新模式,即国内创新药企业通过技术入股,与海外资本一起攒局。尽管颇有资本运作的味道,但这种出海模式既能通过早期管线创造一定价值,又能通过股权,降低自身风险、参与公司决策,并锁定更多远期收益。
2024年出海相关标的:
• 和黄医药:赛沃替尼联合奥希替尼在奥希替尼经治的非小细胞肺癌Ⅱ期数据有望于2024年下半年读出,并递交NDA。索乐匹尼布ESLIM-01 III期研究结果优异,有望持续探索多项自身免疫病。
• 迪哲医药:首个全球注册临床研究——WU-KONG1达到主要研究终点,ORR达53.3%,有望2024年下半年海外NDA。
• 海思科:环泊酚美国Ⅲ期研究显示,与丙泊酚相比,环泊酚药物相关不良事件更少,注射痛显著降低。环泊酚有望2024年下半年海外NDA。
• 荣昌生物:泰它西普多项适应症在海外处于临床三期,箭在弦上。RC88作为MSLN靶点的佼佼者,在2024ASCO展现出不俗的治疗潜力。
• 泽璟制药:ZG005在末线宫颈癌数据优异,ZG006是小细胞肺癌的潜力药物,多款DLL3多抗成功授权。
• 迈威生物:9MW2821在晚期 UC、CC、EC 和 TNBC 中的展现了令人鼓舞的疗效。
• 亚盛医药:APG-2575美国处于临床Ⅲ期, Bcl-2赛道先发优势明显。
风险提示:创新药海外研发的不确定性、海外研发进展低于预期、海外商业化进展不及预期、FDA政策风险等。
一、创新药出海征途开启,广阔天地大有可为
1.1 创新药出海的内在逻辑:医疗支出差距、药品价格差距、药品峰值差距和快速回
笼资金
创新药出海的内在逻辑1:
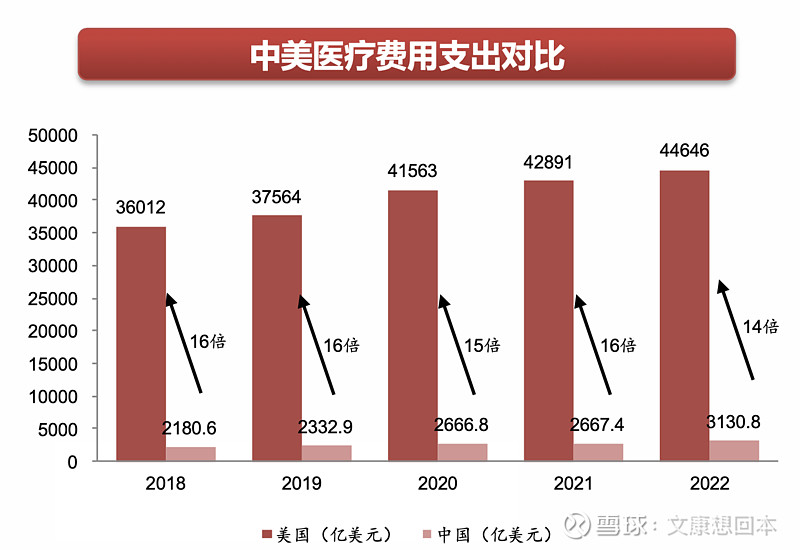
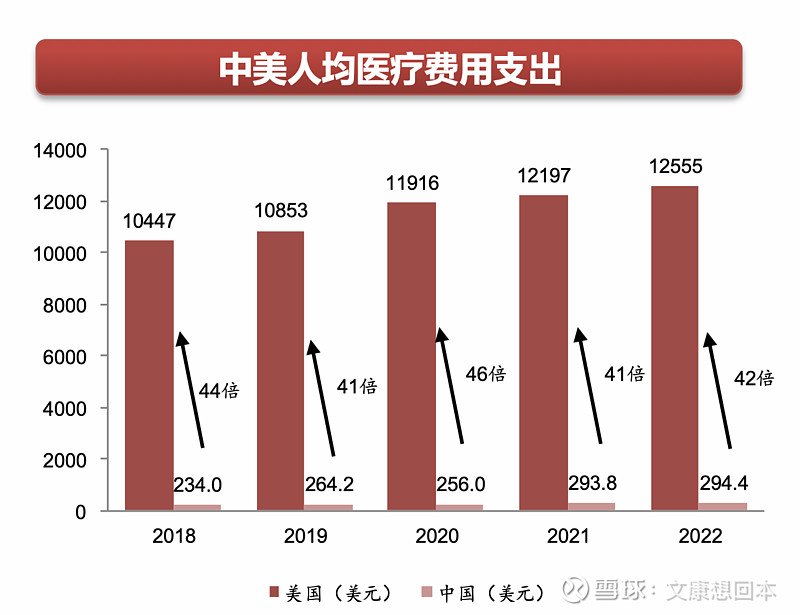
中美医疗支付能力差距显著,海外市场空间巨大。中国人口众多,医疗市场体量庞大,位列全球第二,仅次于美国。2018年,美国总医疗费用支出是中国的16倍。但人均医疗费用远落后于美国,同年美国人均医疗费用支出是中国的44倍。尽管近年来中国医疗行业发展迅速,与国际差距逐步缩小,但冰冻三尺非一日之寒,2022年中美医疗费用支出仍存在14倍的差距,人均医疗费用仍有42倍的差距。
随着国内新药研发实力的快速提升,药物创新性和研发效率的优势受到全球的认可,新药出海对于企业而言势在必行。但出海本无须如此迫切,只是在带量采购、医保谈判、创新药内卷加剧等冲击下,国内创新药企业被迫寻找新的出路和发展空间。考虑到太平洋彼岸的美国市场存在着巨大的市场空间和想象空间,国内企业争相将海外战略付诸实践,加速全球化进程。
销售额出海的内在逻辑2:
中美药品销售额差距逐渐拉大,药品峰值存在巨大差异
泽布替尼是百济神州旗下一款BTK抑制剂。2022年,泽布替尼在美国实现26.4亿元销售额,国内销售额为10亿元,美国和中国市场销售额已拉开差距。而这差距仍在逐步扩大。2023年,泽布替尼美国收入69亿元,中国收入14亿元,差距已达4.9倍,销售额差距的加大反映出中美市场的巨大差异,一方面是中美药品治疗费用的差异,另一方面则是药品峰值的差异。
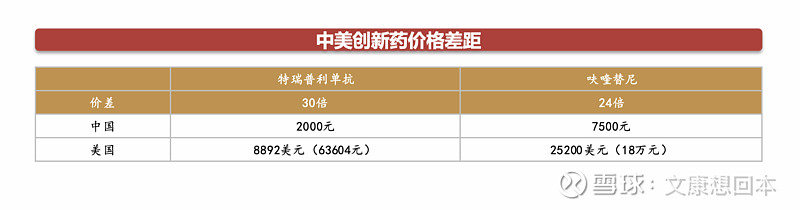
创新药在国际市场上的售价通常更高,多款产品价差超20倍。
君实生物的PD-1抑制剂特瑞普利单抗于2023年10月底获得美国FDA的批准后,在美国的定价为每瓶8892.03美元,折合人民币约63604.69元,这一价格是其在中国市场不足2000元人民币售价的30倍以上。
同样,和黄医药的呋喹替尼(5mg*21粒)于2023年11月初获得美国FDA批准后,在美国市场的定价为25200美元,折合人民币约18.04万元,这是其在中国市场7500多元人民币售价的24倍。
销售额出海的内在逻辑3:
首付款快速回笼资金,寻找伙伴加速海外临床试验
随着生物医药行业投融资频率下降,IPO步伐减缓,在高研发投入的大背景下,部分创新药企业账上资金紧张,“活下去” 成为创新药企业追求的目标之一。
创新药出海,尤其是License out,首付款可助力创新药企业迅速回笼资金。此外,海外临床研究,尤其是Ⅲ期临床研究,研发成本高昂,通过License out授权,可将海外临床研发成本交由海外企业承担,对于创新药企业而言,不但大幅减少了资金压力,更有助于快速推进临床试验。
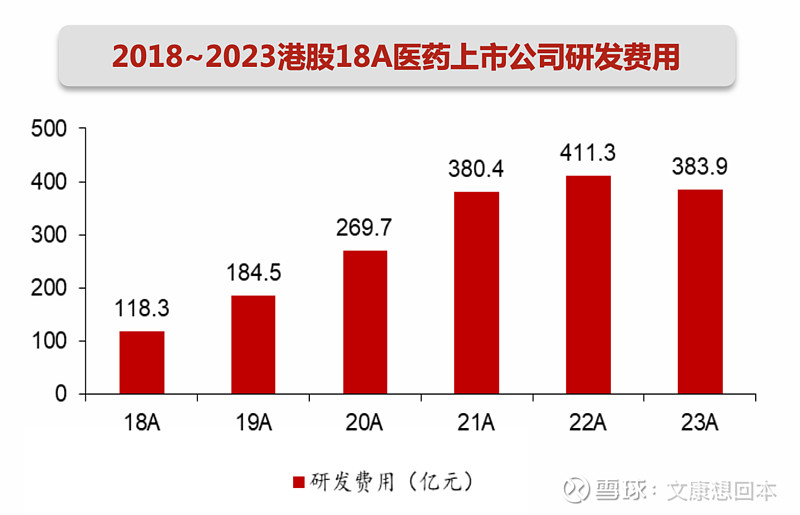
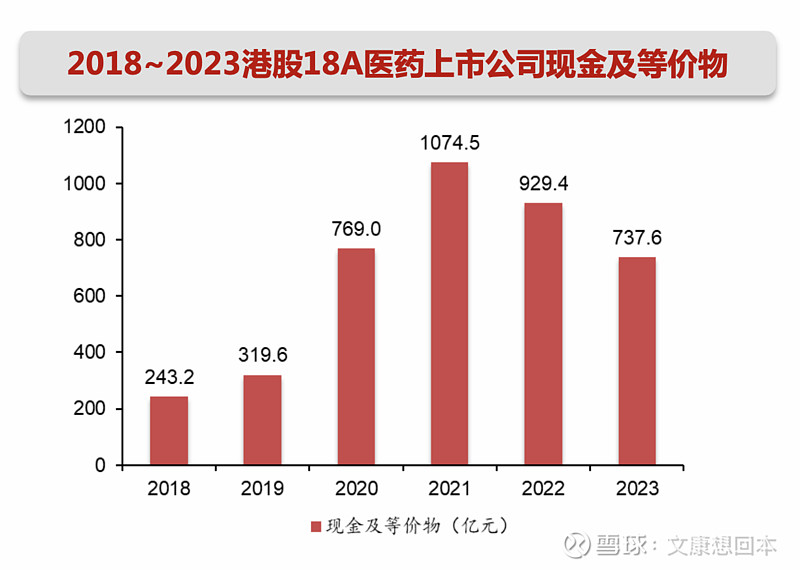
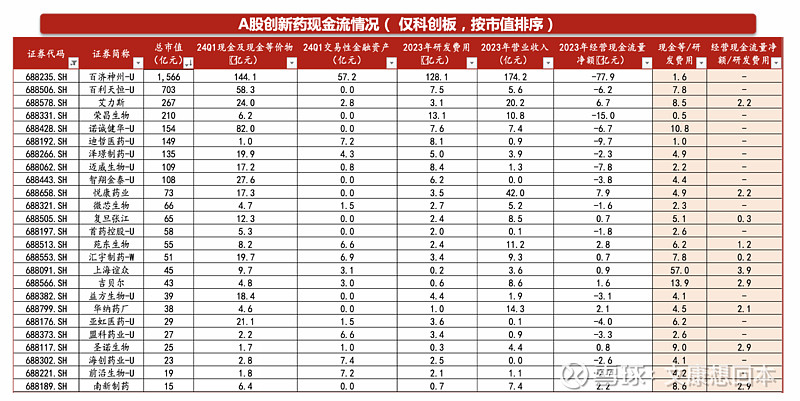
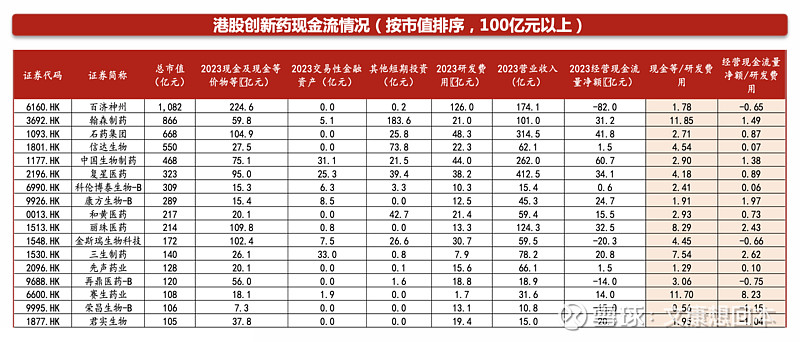
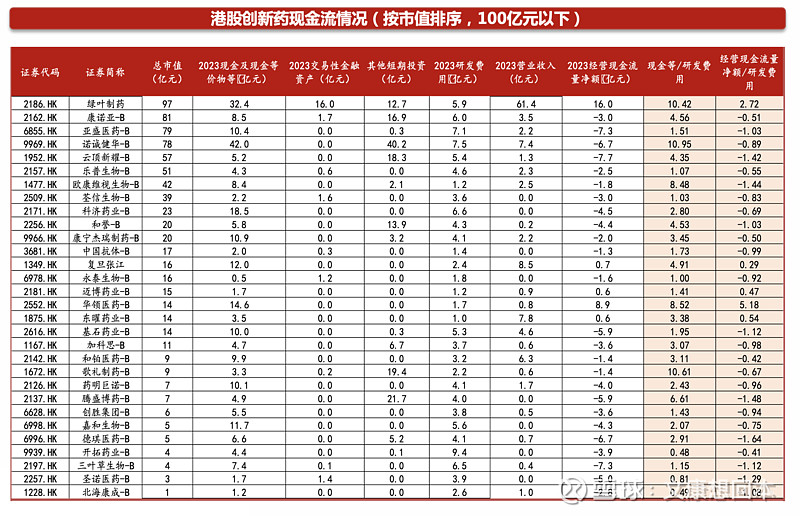
对于尚未有产品商业化收入的科创板Biotech,“现金/研发费用“比值中位数约为4.9。部分Biotech的技术服务收入、授权收入等可以贡献较多现金,比如百利天恒BL-B01D1于2023年底和BMS的重磅合作,将获得8亿美元的首付款和最高5亿美元的或有近期付款。还有部分Biotech处于产品放量元年/初期,2024年产品收入将有明显增长从而弥补现金流,比如迪哲医药的舒沃替尼2023年8月获批上市。
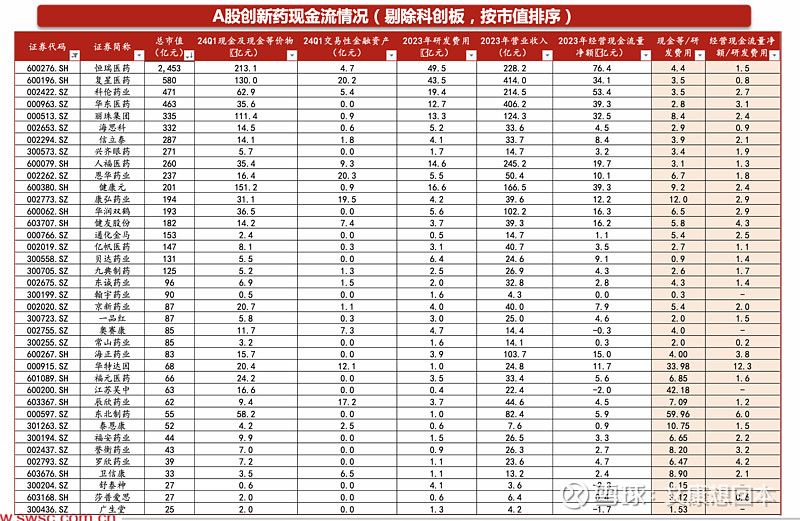
非科创板的A股药企基本具备较稳定的经营现金流量净额,“经营现金流量额/研发费用”比值中位数约为1.8。非科创板的A股药企基本具备自身造血能力,经营现金流稳健。
港股创新药公司“现金等/研发费用“比值中位数约为2.9。港股100亿以上的pharma型公司均具有较充裕的现金(包括现金等价物等)和自身造血能力。Biotech随着管线陆续步入收获期以及出海,近半数公司产生收入。部分Biotech的技术服务收入、授权收入等可以贡献较多现金,比如2023年12月和铂医药与Seagen就MSLN ADC的全球临床开发及商业化订立许可协议,将获得首付款5300万美元。还有部分Biotech处于产品放量元年/初期,产品收入快速增长,比如荣昌生物的泰它西普、维迪西妥单抗。
1.2 创新药出海的三要素:市场潜力、临床数据、先发优势
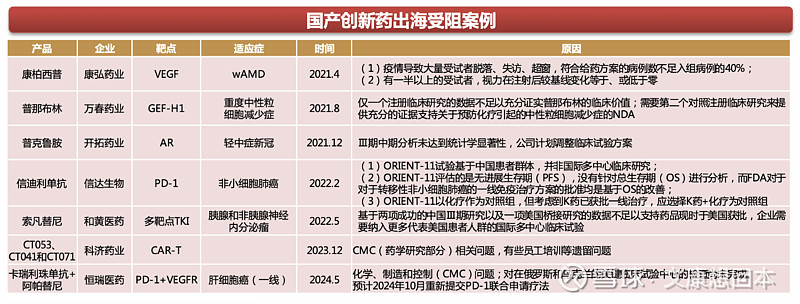
近年来,尽管超50款国产创新药成功出海,并且多款实现了十亿以上的交易。但必须承认的是,国产创新药海外授权从来不是一件容易的事情,尤其是十亿以上的大单,康柏西普、信迪利单抗、索凡替尼等接连受阻。
从受阻原因出发,信迪利单抗、普那布林和索凡替尼均缺少代表美国患者人群的国际多中心临床试验数据;康柏西普的海外试验受疫情影响,大量患者脱落,且一半以上的受试者疗效不及预期;CAR-T和卡瑞利珠单抗因CMC问题,临床研究被暂停。
启示:
1)兼具疗效和安全性的差异化产品;
2)研究和申报程序要全面符合美国药品监管的体系要求,即包含美国患者的临床数据和国际多中心临床试验;
3)FDA规定的CMC相关问题;
4)潜在因素的影响,如口罩导致的现场核查受阻。
创新药出海须满足三要素:
首先,市场空间巨大,可有效解决未满足的临床需求。市场空间的潜力是MNC最在意的问题,如强生携手传奇生物共同推进西达基奥仑赛的商业化,强生预计西达基奥仑赛全球销售额有望达50亿美元。Summit Therapeutics引进康方生物的AK112,并与K药在非小细胞肺癌展开头对头研究。考虑到K药2023年全球销售额达250亿美元,若AK112头对头战胜K药,有望在非小细胞肺癌中斩获不俗的市场份额。
其次,临床有效性及安全性较目前标准疗法优势明显。药物的临床疗效和安全性优势是药品拓展市场的关键,最终反映出医生和患者对药品的信赖度。武田医药引进呋喹替尼,一方面,呋喹替尼疗效优势明显,在末线结直肠癌中mPFS达3.7m,mOS达7.4m,而竞品瑞戈非尼mPFS仅3.2m,mOS仅6.4m;另一方面,呋喹替尼安全性良好,反观瑞戈非尼则存在肝毒性黑框警告。安全性的优势则意味着武田还能探索呋喹替尼联用IO的疗法,有望在多项适应症中展露锋芒。
最后,临床进展居前,先发优势明显。药品的先发优势对于药物的放量至关重要。根据《2020年度中国抗肿瘤新药临床研究评述》,全球范围内同一靶点药物,首个上市产品可以获得45%的市场份额,第二至第四个上市的产品分别可以获得27.9%、14%以及11.3%的市场份额。如辉瑞引进科伦博泰的SKB264是全球研发进展前三的Trop2 ADC, Summit Therapeutics引进康方生物的AK112为全球首款获批上市的PD-1/VEGF双抗。
1.3 自主出海和License out日臻成熟,联手出海发展方兴未艾
出海主要分为自主出海、License out(借船出海)和联手出海。
自主出海即中国药企凭借自身的团队在海外国家和地区开展临床试验,申报上市,获批后展开销售。考虑到自主出海的高风险,部分企业采用联手出海的模式,即通过与海外药企联合开发,分担成本并分享收益。
License out则是中国药企将自身产品的海外权益或全球权益出手许可给以欧美跨国药企为代表的制药企业,获得首付款和里程碑费用。海外药企接过接力棒后,负责海外市场的临床开发、申报上市、生产及销售工作。
目前License out是出海的主要模式,企业可根据自身的战略规划和实力,在不同阶段选择不同的模式,或采用多种模式并行的方式。
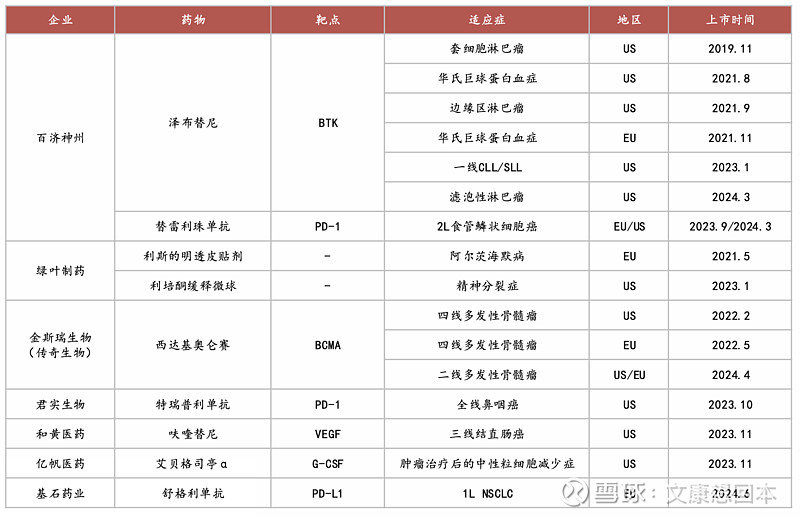
国产创新药海外上市品种
西达基奥仑赛销售额持续攀升
传奇生物:2022年2月底,西达基奥仑赛通过美国食品药品监督管理局(FDA)批准上市,成为首款获得美国FDA批准的中国CAR-T细胞疗法。2022年5 月 26 日,传奇生物宣布欧盟委员会(EC)已授予 CARVYKTI®(西达基奥仑赛)附条件上市许可,用于治疗既往接受过至少三种治疗,包括免疫调节药物、蛋白酶体抑制剂和抗 CD38 抗体,并且末次治疗出现疾病进展的复发或难治性多发性骨髓瘤(R/R MM)成人患者。
2024年4月22日,欧盟委员会(EC)已批准西达基奥仑赛(cilta-cel,商品名:CARVYKTI®)用于治疗复发和难治性多发性骨髓瘤成人患者。这些患者既往至少接受过一线治疗,包括一种蛋白酶体抑制剂(PI)和一种免疫调节剂(IMiD。
2017 年 12 月,强生子公司 Janssen 与传奇生物签订了独家全球许可和合作协议,以开发和商业化西达基奥仑赛,这笔交易首付款达 3.5 亿美元。在美国,两家公司的权益按 50:50 分成。2023年,西达基奥仑赛全球销售额达4.98亿美元。
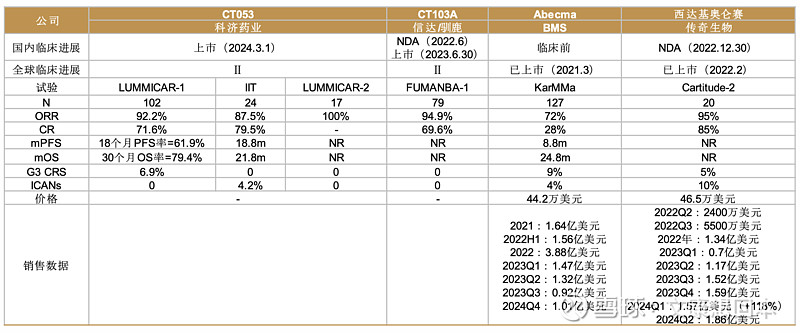
国产创新药美国注册性临床梳理
出海密集兑现,重视海外商业化成绩单催化,持续关注出海里程碑进展
关注重要Ⅲ期品种临床数据读出和上市申请节点。22家中国药企的25个创新药已在美国开展注册性临床试验(剔除新冠药物和美国NDA/临床失败药物),重磅品种包括SKB264、依沃西单抗、泰它西普、舒沃替尼、赛沃替尼、伏美替尼等。
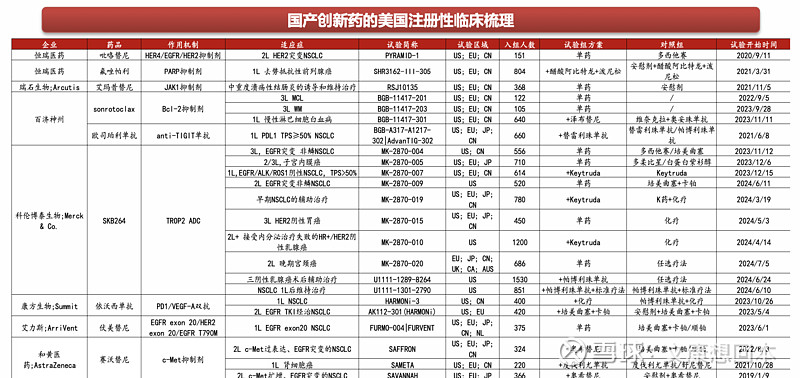
国产创新药美国3期临床梳理
出海密集兑现,重视海外商业化成绩单催化,持续关注出海里程碑进展
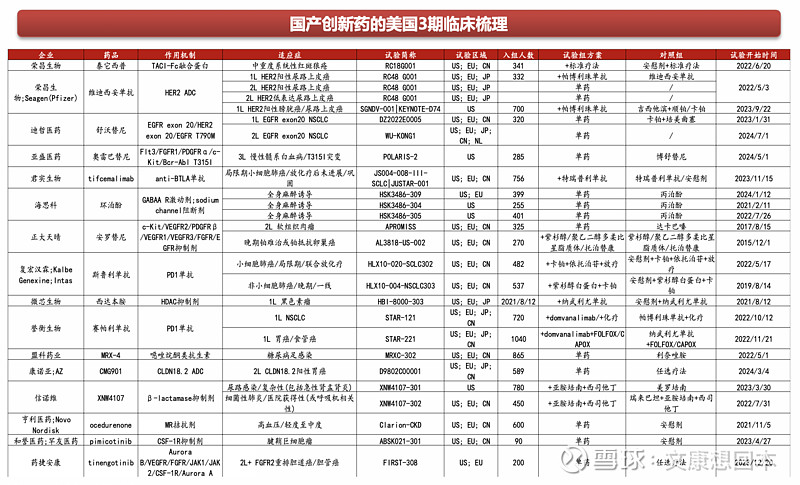
FDA特殊审评通道简介
FDA为鼓励罕见病和严重疾病创新药的开发制定了一系列的特殊审评通道,包括:孤儿药、快速通道、加速审批、优先审评和突破性疗法认定。
孤儿药(Orphan):美国国会于 1983 年推出《孤儿药法案》(ODA),提出一系列推动罕见病药物研发的政策,如税收抵免(临床研发成本的25%),为孤儿药项目提供研究资助,免除NDA/BLA费用(~ 300 万美元),上市后7年市场独占期等。此外,临床试验期间药企还可与FDA 对临床试验的设计进行紧密沟通,缩短 IND 和 NDA 的时间。
快速通道(FAST TRACK):美国于1988年推出快速审评通道计划,以“促进治疗严重疾病和满足未满足的医疗需求的药物的开发和加快审查”。在快速通道流程下,制药公司在I期试验后与FDA 讨论II期试验设计,如果成功,II期试验(而非III期)将足以证明药物的安全性和有效性。根据该计划,FDA 可以持续审查临床试验产生的证据(滚动审查)。
加速审批(Accelerated approval):加速批准使 FDA根据替代指标而非临床终点来判断药物的疗效。衡量药物对患者生存的影响可能需要较长的试验时间和随访数据(特别是对于 5 年和10 年生存率较高的癌症类型,例如前列腺癌或乳腺癌)。相比之下,替代终点(例如肿瘤缩小或进展)可以更快地观察到。因此,加速审批计划通过缩短临床试验持续时间并启用较少入组患者的试验设计来测量替代终点,从而加快了药物开发。有研究显示,获得加速审批的癌症药物比那些获得标准FDA批准的药物更早进入市场,平均提前了3.9年。另外,研究表明使用PFS(无进展生存期)和肿瘤反应率这些替代终点可以分别将临床试验时间缩短11个月和19个月。
优先审评(riority review):标准 NDA 的审核周期为10 个月(2002 年为 12 个月),而优先NDA 的审核周期为6个月。与其他特殊指定不同,审查途径由FDA收到NDA和补充适应症材料后自行判定。
突破性疗法认定(Breakthrough therapy designation):2012 年,美国国会引入了新的 FDA 审查途径:突破性疗法。该认定提供与 FDA 高级人员更早(I期)和更频繁的会议,以指导有效的药物开发和监管审查流程。此外,突破性疗法指定的药物可以获得快速通道计划的所有好处。
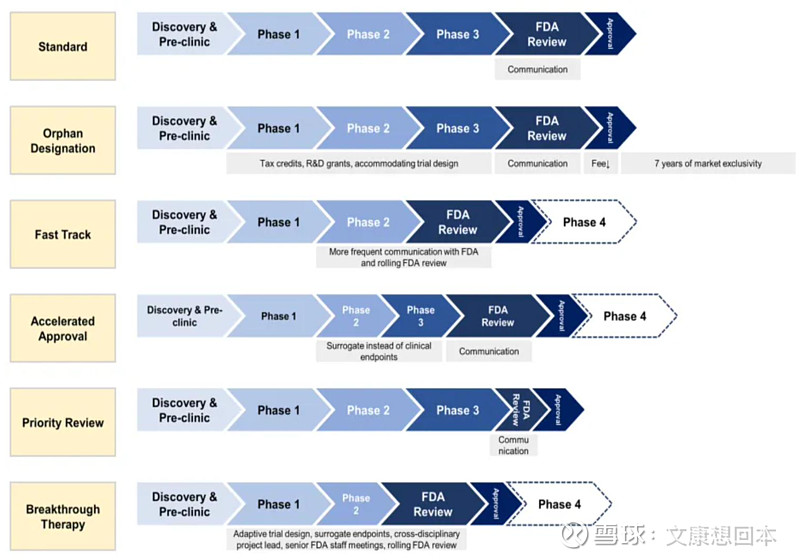
获得美国突破性疗法和加速批准的国产创新药
出海密集兑现,重视海外商业化成绩单催化,持续关注出海里程碑进展
16款国产创新药获得美国突破性疗法认定,其中泽布替尼还获得了加速批准。
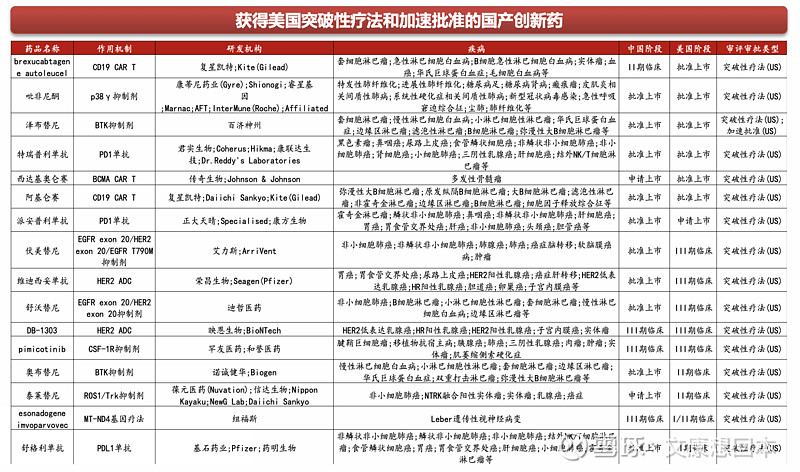
2024年截止7月8日,超15款国产创新药获得美国快速通道认定,主要为ADC、PROTAC、多抗、核药、溶瘤病毒等前沿疗法,适应症主要为患者人数较少、现有疗法不能满足临床需求的适应症。
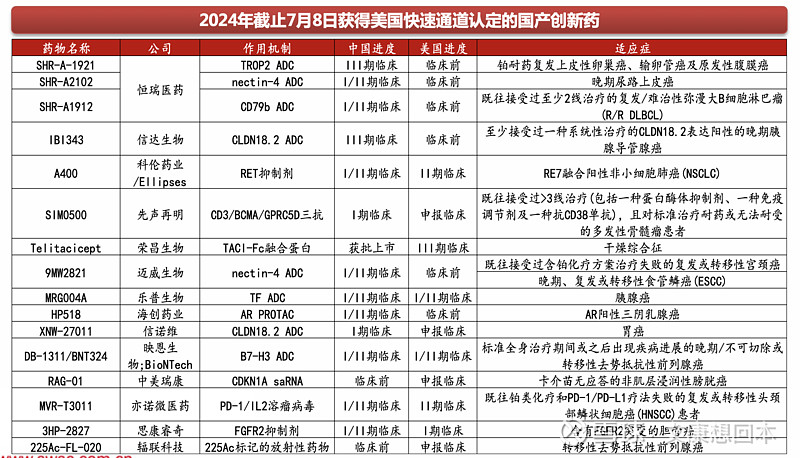
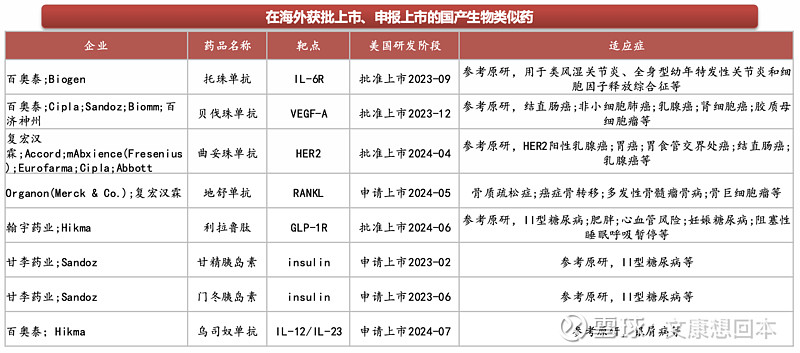
在美国获批上市、申报上市的国产生物类似药
2023年美国市场生物类似药销售收入约72.5亿美元,是全球最大的生物类似药市场。截至2024年2月,美国以351(K)途径申报的生物类似药获批总数量和上市数量已经分别达到了46个和38个。从产品类型看,共涉及维持治疗、抗肿瘤、自体免疫、胰岛素和眼科五大领域的14个分子,其中数量最多的阿达木单抗共有10个生物类似药获批,其中9个已经上市,成为当下竞争最为激烈的产品,数量最少的阿法依泊汀只有1个生物类似药获批并上市。
中国生物类似药抢滩美国市场,在美国获批上市、申报上市的国产生物类似药已有8款,均有2023年后获批/申报。百奥泰、甘李药业等是我国生物类似药出海先锋,百奥泰已有2款类似药在美国获批上市、还有3款类似药处于国际3期临床阶段/接近申报上市,甘李药业两款三代胰岛素也已于2023年在美国申报上市,市场前景广阔。
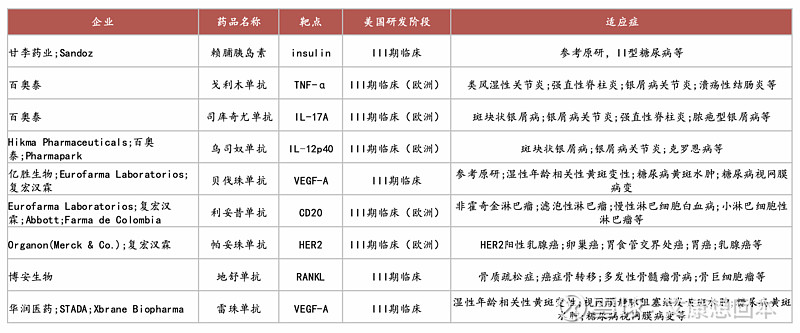
在海外处于临床Ⅲ期的国产生物类似药
1.5 License out:渐入佳境,交易数量和交易金额攀升
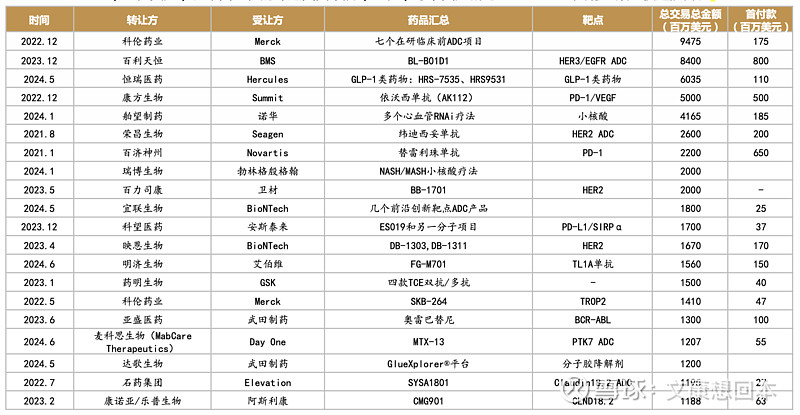
License out大手笔频现,不断刷新交易金额
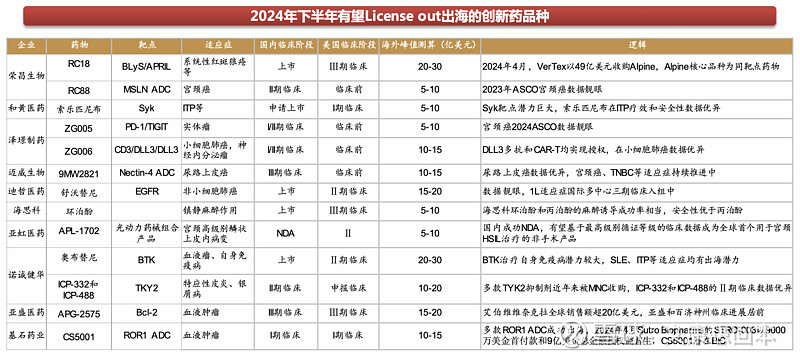
License out大手笔频现,不断刷新交易金额。随着国内新药创制水平的不断提升,国产创新药的国际认可度稳步上升,国产创新药License out金额持续攀升。据不完全统计,超过30个License out项目总交易金额超5亿美元,其中超20亿美元的项目包括科伦药业的七个ADC项目(94.7亿美元)、百利天恒的BL-B01D1(84亿美元)、康方生物AK112(50亿美元)、荣昌生物的纬迪西妥单抗(26亿)等,国产新药License out交易金额正快速提升。
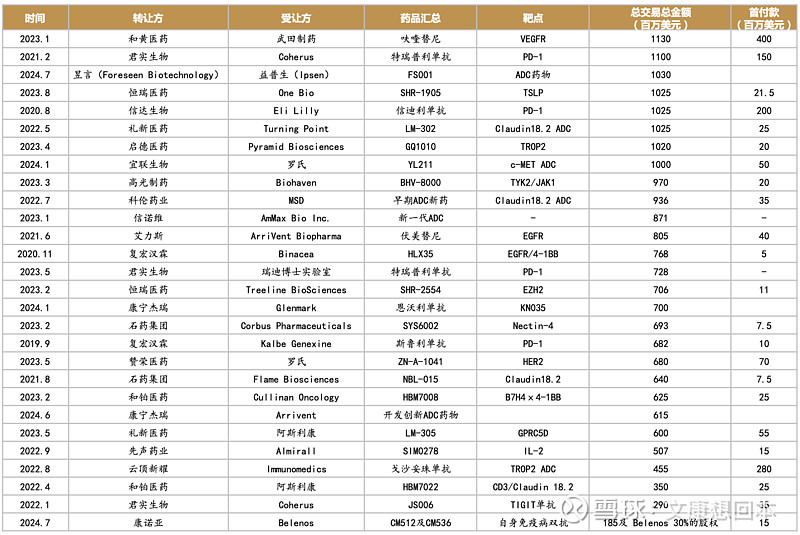
2024年上半年License out梳理
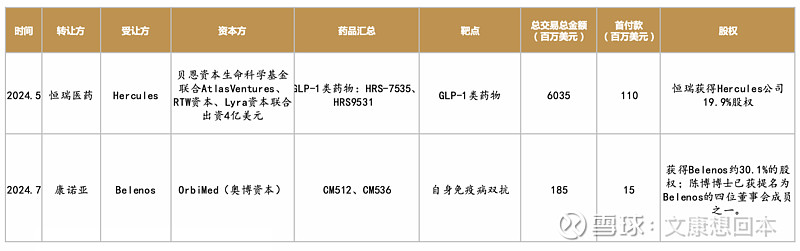
与资本合作出海,License out新趋势
• 这种模式跟传统License out不同,在新模式下,国内创新药企业通过技术入股,与海外资本一起攒局。“找不到MNC就先卖给中间商,没有中间商就和海外资本一起创造中间商。”
• 尽管颇有资本运作的味道,但不能否认这种出海模式的好处,既能通过早期管线创造一定价值,又能通过股权,降低自身风险、参与公司决策,并锁定更多远期收益。
二、2024年下半年出海潜力管线分析
2.1 2024年下半年出海相关标的
2024年下半年有望出海的创新药品种
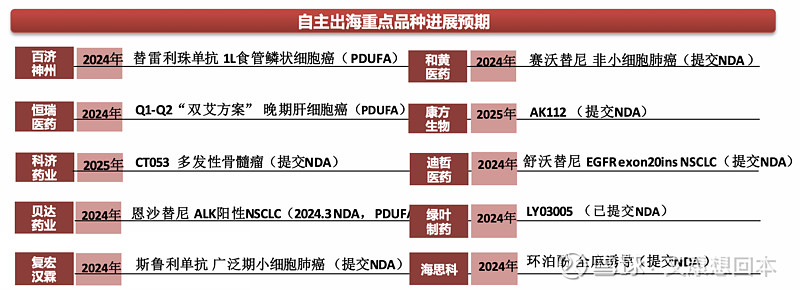
多款国产创新药正在海外开展临床试验,我们认为,放眼2024年下半年及2025年,以下品种有望迎来关键的海外催化:
和黄医药:赛沃替尼联合奥希替尼在奥希替尼经治的非小细胞肺癌Ⅱ期数据有望于2024年下半年读出,并递交NDA。该海外临床由阿斯利康负责推进,鉴于奥希替尼2023年全球销售额达57.9亿美元,奥希替尼耐药后线市场庞大。
康方生物:AK112海外两项Ⅲ期研究正在推进中,包括EGFR-TKI治疗失败的非小细胞肺癌和1L 非小细胞肺癌。鉴于AK112海外头对头K药,一旦成功,有望替代K药在一线NSCLC的地位。
迪哲医药:首个全球注册临床研究——WU-KONG1达到主要研究终点,ORR达53.3%,有望2024年下半年海外NDA。
海思科:环泊酚美国Ⅲ期研究显示,环泊酚麻醉诱导成功率不劣于丙泊酚。与丙泊酚相比,环泊酚药物相关不良事件更少,注射痛显著降低。
2.2 沙海淘金,具体标的分析
荣昌生物:泰它西普多项适应症步入国际三期
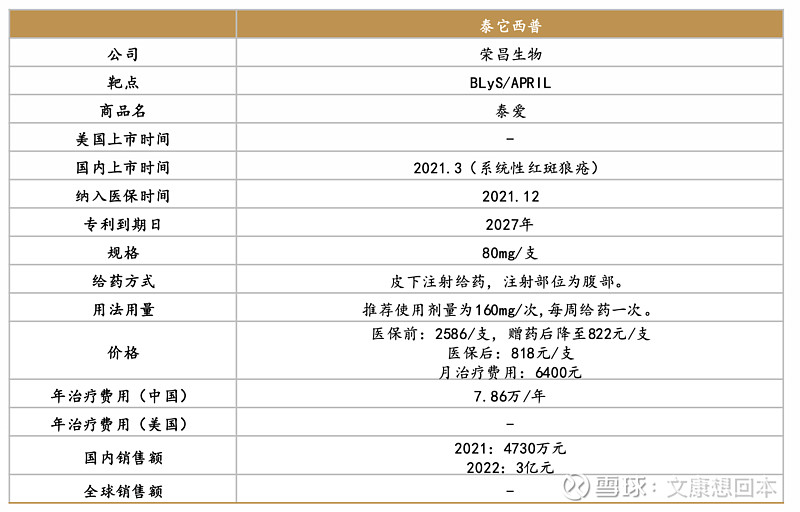
泰它西普是全球首个靶向B淋巴细胞刺激因子(BLyS)和增殖诱导配体(APRIL) 双靶点机制药物,由荣昌生物研发。2021年3月,泰它西普获批上市,用于治疗系统性红斑狼疮。
泰它西普多项适应症在海外处于临床三期,箭在弦上。2022年11月18日,荣昌生物宣布,美国食品药品监督管理局(FDA)已同意泰它西普在美国开展IgA肾病适应症的Ⅲ期临床试验。2022年4月,荣昌生物泰它西普在美国启动首相三期临床REMESLE-1,治疗系统性红斑狼疮。2024年6月13日,荣昌生物在Clinicaltrials.gov网站上注册了泰它西普两项美国三期临床,分别用于治疗中至重度系统性红斑狼疮(SLE)、全身型重症肌无力(gMG)。 2023年12月,泰它西普治疗原发性干燥综合症(pSS)的三期临床获得FDA许可。
荣昌生物:RC88在多项实体瘤中展现出治疗潜力
RC88是由荣昌生物自主研发、具有first-in-class潜质的靶向MSLN的ADC药物,用于铂耐药复发性上皮性卵巢癌,输卵管癌和原发性腹膜癌,已获 FDA 快速通道资格认定,II期研究已经在美国和中国启动。
由中国医学科学院肿瘤医院石远凯教授牵头的RC88在卵巢癌、非鳞状非小细胞肺癌和宫颈癌患者中的疗效和安全性:一项首次人体I/II期临床研究的结果,在2024年ASCO大会上以壁报的形式发布。
截至2024年2月21日,170例晚期实体瘤患者入组。剂量递增阶段完成,2.0 mg/kg和2.5 mg/kg Q3W剂量扩展到II期。
在卵巢癌(OC)队列中,截至2024年3月22日,2.0mg/kg组共有31例患者接受了2至4线既往治疗,可进行疗效评估。其中ORR为45.2%(cORR 41.9%),中位DoR为8.02个月。
在非鳞状非小细胞肺癌(NSCLC)队列中,16例可评估患者的ORR为31.3%(cORR 25%),上述MSLN高表达患者的ORR、cORR、中位PFS和中位DoR分别为41.7%、33.3%、6.87个月和9.13个月。
在宫颈癌(CC)队列中,18例可评估患者的ORR为33.3%(cORR 27.8%)。在接受≥2线治疗的12例患者中,ORR为41.7%(cORR 33.3%)。
目前,铂耐药卵巢癌标准治疗方案化疗的ORR为12%,这项研究的ORR为45.2%(cORR 41.9%),令人鼓舞的结果显示了RC88在显著改善MSLN表达的晚期实体瘤患者预后方面的潜力。
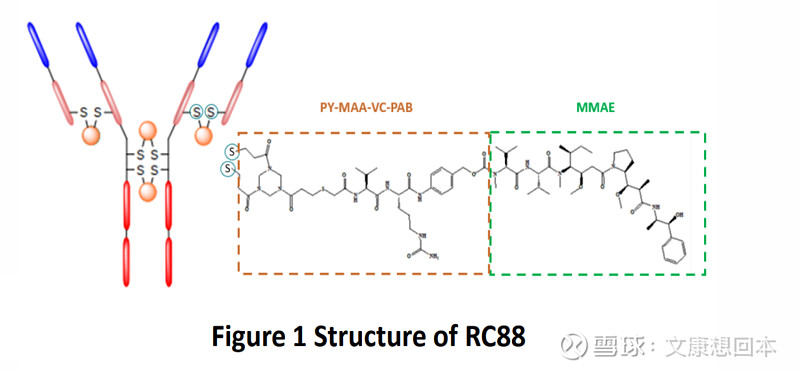
和黄医药:索乐匹尼布ESLIM-01 III期研究优异
索乐匹尼布是一种用于治疗血液恶性肿瘤和免疫性疾病的新型、高选择性的脾酪氨酸激酶 ("Syk") 口服抑制剂。
ESLIM-01 III期研究的结果于2024年《柳叶刀·血液病学》发表。在既往接受过四线或以上治疗的患者中,索乐匹尼布组的持续应答率为 47.7%,而安慰剂组则为 0% (p <0.0001)。此外,大部分患者既往曾接受过TPO/TPO-RA治疗。索乐匹尼布组的患者中74.6%的患者曾接受过TPO/TPO-RA治疗,对该亚组的分析显示,索乐匹尼布组的持续应答率为46.8%,而安慰剂组则为零 (p <0.0001)。在接受索乐匹尼布治疗的患者中,25.4% 出现 3 级或以上TEAE,而安慰剂组则为 24.2%。
中国国家药品监督管理局已将索乐匹尼布用于此适应症纳入突破性治疗品种,并于2024年1月受理其新药上市申请并纳入优先审评。一项于美国开展的剂量探索研究正在计划中 (NCT06291415)。和黄医药亦已于 2024 年 3 月在中国启动索乐匹尼布用于治疗温抗体型自身免疫性溶血性贫血 (wAIHA) 成人患者的II/III 期研究的注册阶段 (NCT05535933)。和黄医药目前保留索乐匹尼布在全球的所有权利。
泽璟制药:ZG005在宫颈癌中疗效优势显著
ZG005是重组人源化抗PD-1/TIGIT双特异性抗体粉针剂,为创新型肿瘤免疫治疗生物制品,注册分类为1类,有望用于治疗多种实体瘤。根据公开查询,ZG005是全球率先进入临床研究的同靶点药物之一,目前全球范围内尚未有同类机制药物获批上市。
ZG005在晚期实体瘤患者中的剂量递增、耐受性、安全性、药代动力学和多队列扩展的I/II期临床研究数据及最新进展:
截至2024年4月16日,ZG005-001项目剂量递增阶段已完成,共入组32例受试者;剂量扩展阶段正在进行中,已入组47例受试者。79例受试者中,43%的受试者既往曾接受过至少两线抗肿瘤药物系统治疗,48%的受试者既往接受过PD-1/PD-L1抑制剂治疗。
有效性方面,可评估疗效的21例宫颈癌受试者(3 mg/kg组1例、10 mg/kg组12例、20 mg/kg组8例)中,有2例完全缓解(CR)、7例部分缓解(PR)和8例疾病稳定(SD);客观缓解率(ORR)为43%(9/21),疾病控制率(DCR)为81%(17/21)。特别是目标剂量20 mg/kg组的客观缓解率(ORR)达到63%。9例获得客观缓解(CR/PRs)的宫颈癌受试者中,2例既往接受过PD-1/PD-L1抑制剂治疗。1例3 mg/kg组宫颈癌受试者,其肿瘤病灶自首次给药后持续缩小,现已接受ZG005治疗超过77周,靶病灶总径较基线缩小60.9%。11例宫颈癌受试者在首次肿瘤评估时即出现肿瘤缩小。截至数据截止日期,有16例受试者仍正在接受治疗。
安全性方面,63.3%(50/79)的受试者出现了与治疗相关的不良事件(TRAEs),绝大多数严重程度为1或2级。
泽璟制药:ZG006结构新颖,剑指实体瘤
ZG006是泽璟制药旗下一种针对CD3及两个不同DLL3表位的三特异性抗体。ZG006的抗DLL3端与肿瘤细胞表面不同DLL3表位相结合,抗CD3端结合T细胞。ZG006衔接肿瘤细胞和T细胞,将T细胞拉近肿瘤细胞,从而利用T细胞特异性杀伤肿瘤细胞。
结合两个不同表位的DLL3,表现出对DLL3很强的结合亲和力。通过靶向DLL3和CD3将肿瘤细胞和T细胞结合,用于T 细胞介导的肿瘤细胞杀伤。对于DLL3不同表达量的肿瘤细胞均有较好的杀伤效果。
多款DLL3在研企业成功授权。2024年1月,默沙东宣布以6.8亿美元收购Harpoon Therapeutics。2023年11月3日,传奇生物全资子公司传奇爱尔兰与诺华就靶向DLL3 CAR-T疗法签订了独家全球许可协议,诺华获得自体CAR-T细胞疗法候选药物LB2102的全球独家权利,同时可以将其TCharge平台应用于以上候选药物的生产;而传奇生物将获得1亿美元的预付款、潜在高达10.1亿美元的里程碑付款及分级特许权使用费。
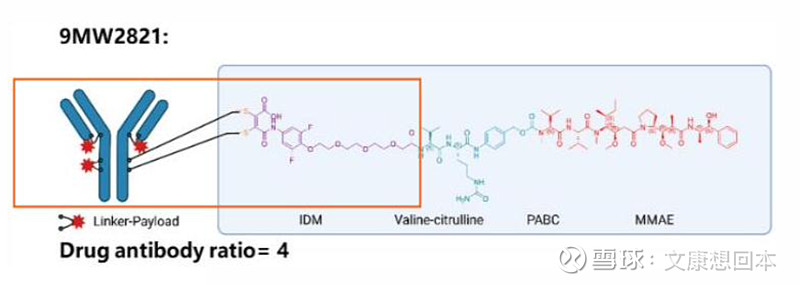
迈威生物:9MW2821在晚期 UC、CC、EC 和 TNBC 中的展现了令人鼓舞的疗效
9MW2821为迈威生物靶向Nectin-4的定点偶联ADC新药,能够将毒素MMAE递送至表达Nectin-4的细胞。
在2024ASCO上,迈威生物披露了9MW2821治疗尿路上皮癌 (UC)、宫颈癌 (CC)、食管癌 (EC) 和三阴乳腺癌的Ⅱ期数据。37例UC患者、53例CC患者、39 例EC患者和20例TNBC患者接受了1.25mg/kg 剂量的治疗,并进行了肿瘤评估。既往中位治疗线数为2(范围为1-4)。
37例可评估疗效的UC患者中:客观缓解率 (ORR) 和疾病控制率 (DCR) 分别为62.2%和91.9%,中位无进展生存期 (mPFS)为8.8个月,中位总生存期(mOS)为14.2个月。
53例可评估疗效的CC患者中:51%受试者既往接受过含铂双药化疗及贝伐单抗治疗,58%受试者既往接受过含铂双药化疗及免疫检查点抑制剂治疗,ORR和DCR分别为35.8%和81.1%,mPFS为3.9个月,mOS尚未达到。Nectin-4 肿瘤细胞染色强度3+的患者中,39例可评估疗效的患者ORR为43.6%。
39例可评估疗效的EC患者中:ORR和DCR分别为23.1%和69.2%,mPFS为3.9个月,mOS为8.2个月;其中37例接受过铂类化疗及免疫治疗。
20例可评估疗效的局部晚期或转移性 TNBC 患者中:ORR 和 DCR 分别为 50.0% 和 80.0%,mPFS为5.9个月,mOS尚未达到;其中,1例完全缓解(CR)患者已持续治疗20个月,目前仍持续完全缓解。
数据显示,9MW2821在晚期UC、CC、EC和TNBC中的展现了令人鼓舞的疗效。其在安全性方面也显示出了充足的耐受性。
迪哲医药:舒沃替尼有望于2024下半年在美国提交NDA
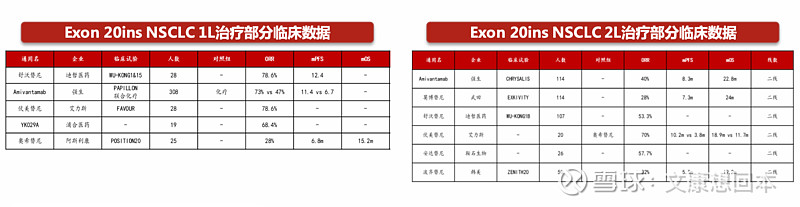
舒沃替尼2L适应症有望于2024下半年在美国提交NDA。首个国际多中心注册临床研究“悟空1B”(WU-KONG1B)主要研究结果,舒沃替尼针对经治的EGFR ex20ins突变非小细胞肺癌(NSCLC)患者最佳客观缓解率(ORR)达53.3%。
舒沃替尼1L适应症国际多中心三期临床入组中 ,有望2025年申报上市。2023年ESMO,舒沃替尼针对EGFR exon20ins突变晚期NSCLC初治患者的全球多中心I/II期研究“悟空1”(WU-KONG1)和中国研究者发起的II期研究“悟空15”(WU-KONG15)的汇总分析显示,截至2023年9月15日,28例患者纳入疗效分析,57例患者纳入安全性分析,研究者评估100%观察到靶病灶缩小,确认的ORR高达78.6%,其中300mg组mPFS为12.4个月,展现出强效持久的抗肿瘤活性,同类最佳潜力凸显。
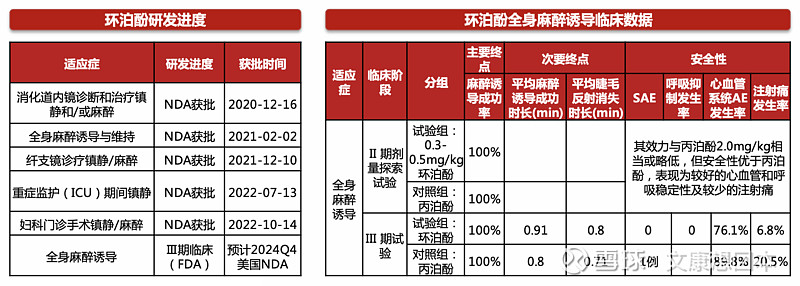
海思科:环泊酚预计2024年Q4在美国提交NDA
海思科环泊酚国内已有5项适应症获批上市,1项适应症处于美国临床Ⅲ期且预计2024年Q4在美国提交NDA。海思科独家新药环泊酚是一种 GABAA受体激动剂,作用机制与丙泊酚相似。通过作用于 GABAA受体介导的氯离子通道,增加电流的传导,引起神经元的超极化,抑制中枢神经系统,产生镇静麻醉作用。
临床Ⅱ期和Ⅲ期试验数据:海思科环泊酚和丙泊酚的麻醉诱导成功率相当,安全性优于丙泊酚。有效性:海思科环泊酚和丙泊酚的麻醉诱导成功率相当,在理想剂量组均为100%,但平均麻醉诱导成功时长和平均睫毛反射消失时长要略长于丙泊酚。安全性:海思科环泊酚明显好于丙泊酚,不良反应、严重不良反应、呼吸抑制、血压下降以及注射痛发生率均低于丙泊酚。
亚虹医药: APL-1702 NDA获受理,计划向FDA递交Ⅲ期临床试验申请
APL-1702是集药物和器械为一体的光动力治疗产品,作为非创伤性的新疗法,治疗过程简单,易于操作,不影响患者日常生活,有望让HSIL患者免除手术治疗的痛苦和副作用,特别针对育龄期女性,能够减少因手术治疗对未来生育带来的不利影响。
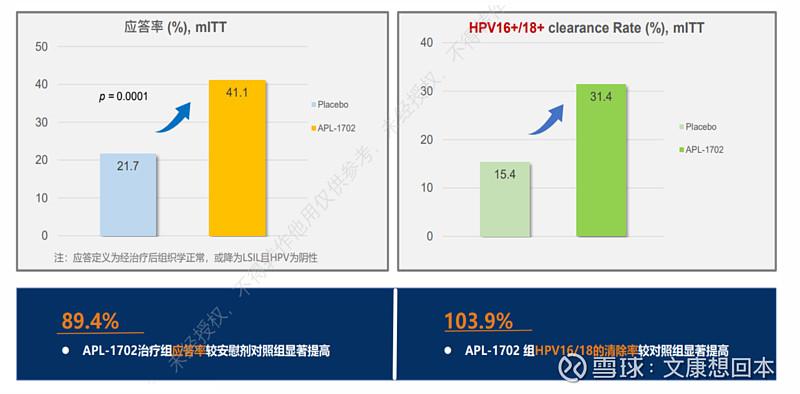
2024年5月,亚虹医药宣布APL-1702(通用名:盐酸氨酮戊酸己酯软膏光动力治疗系统)拟用于治疗18岁及以上排除原位癌的经组织学证实的宫颈高级别鳞状上皮内病变(High-Grade Squamous Intraepithelial Lesion, HSIL)患者的上市申请获得受理。
2023年9月,APL-1702 用于治疗HSIL的前瞻、随机、双盲、安慰剂对照的国际多中心Ⅲ期临床试验达到主要研究终点,APL-1702有望基于最高级别循证等级的临床数据成为全球首个用于宫颈HSIL治疗的非手术产品。
亚虹医药计划就APL-1702向 FDA 递交Ⅲ期临床试验申请。
诺诚健华:两款TYK2抑制剂授权预期高,自身免疫病市场潜力巨大
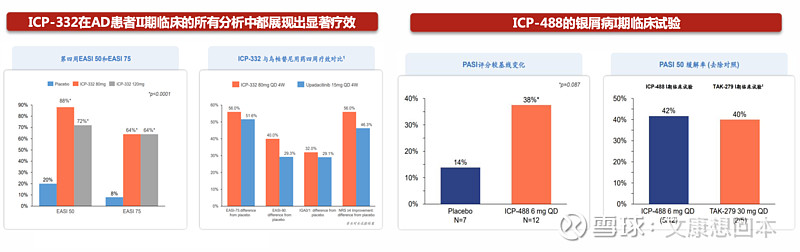
ICP-332是TYK2-JH1抑制剂,对TYK2具有强效抑制活性,比JAK2的选择性高达约400倍。2023年12月18日,ICP-332在中重度特应性皮炎成年患者中进行的Ⅱ期随机、双盲、安慰剂对照研究中取得了积极的临床试验结果。ICP-332在80mg组及/或120mg组中4周时达到了多个有效性终点,包括EASI(湿疹面积和严重程度指数)50、EASI 75、EASI 90(EASI评分较基线改善≥50%,75%,90%)等。EASI评分较基线变化百分比(衡量特应性皮炎的皮损面积和严重程度)在每天一次80毫克时达到78.2%,在每天一次120毫克时达到72.5%,与安慰剂的216.7%相比,具备显著的统计学差异(p<0.0001)。EASI 75在80毫克和120毫克剂量下分別达到64%/64%,而安慰剂组为8%(p<0.0001)。
ICP-488是TYK2-JH2 抑制剂。截至2024年3月28日,公司已完成ICP-488的I临床试验,在健康受试者和中重度慢性斑块型银屑病患者中评估了ICP-488的PK特性和安全性,并在银屑病患者中展现了初步有效性。在接受治疗4周的患者中,ICP-488展示了有效性。在每日一次6毫克剂量组中,银屑病面积和严重程度指数(PASI)评分较基线变化百分比的最小二乘均值较安慰剂组(13.8%)相比改善了23.7%,具有统计学意义。6毫克组PASI 50的应答率较安慰剂组(0%)改善42%。
亚盛医药:APG-2575美国处于临床Ⅲ期,先发优势明显
APG-2575是亚盛医药自主研发的新型口服Bcl-2选择性抑制剂,通过选择性抑制Bcl-2蛋白,恢复癌细胞的正常凋亡过程,从而达到治疗肿瘤的目的。APG-2575在多种血液肿瘤和实体瘤治疗领域具备广阔的单药和联合治疗潜力。
目前,APG-2575正在开展三项注册临床研究,分别为治疗复发/难治性慢性淋巴细胞白血病/小淋巴细胞淋巴瘤(R/RCLL/SLL ) 的关 键 注 册 II 期 临 床 研 究 (APG2575CC201 ) ; 治 疗 经 治CLL/SLL 患 者 的 全 球注 册 III 期 临 床 研 究(APG2575CG301);以及治疗初治CLL/SLL患者的全球注册III期临床研究(APG2575CC301)。
目前,APG-2575共获5项美国FDA授予的孤儿药资格认证(ODD),用于治疗华氏巨球蛋白血症(WM)、慢性淋巴细胞白血病(CLL)、多发性骨髓瘤(MM)、急性髓系白血病(AML)、以及滤泡性淋巴瘤(FL)。
全球仅一款Bcl-2抑制剂上市,为艾伯维的维奈克拉,全球销售额超过22亿美元,亚盛医药的APG-2575临床进展居前,先发优势明显。
基石药业:多款ROR1 ADC实现授权,靶点关注度高
CS5001是一款以受体酪氨酸激酶样孤儿受体1(receptor tyrosine kinase-like orphan receptor 1, ROR1)为靶点的抗体偶联药物(ADC)。CS5001具有独特的设计,使用肿瘤特异激活的吡咯并苯二氮卓(pyrrolobenzodiazepine, PBD)前毒素载荷(Payload)和连接子(linker)。
在2024ASCO披露了CS5001的Ⅰ期数据:截至海报数据截止日,已完成1a期前九个剂量水平(7至156 μg/kg)的剂量限制性毒性(DLT)的评估;未观察到DLT,且未达到最大耐受剂量(MTD)。已观察到的药物相关不良事件大多为1级或2级(评价标准NCI-CTCAE v5.0),表明CS5001针对多线经治的晚期实体瘤和淋巴瘤均能表现出良好的耐受性和安全性。药代动力学数据表明CS5001的暴露量与剂量成正比,且ADC和总抗体的暴露量相似,表明CS5001 ADC在血液循环中具有出色的稳定性。
已在多种实体瘤(评价标准RECIST v1.1)和血液肿瘤(评价标准Lugano 2014)中观察到CS5001令人鼓舞的抗肿瘤活性:
霍奇金淋巴瘤:从第5剂量水平(50 μg/kg)起观察到客观缓解;9例可评估的患者中有1例达到完全缓解(CR)以及4例达到部分缓解(PR),客观缓解率(ORR)为55.6%。
弥漫大B细胞淋巴瘤(DLBCL):从第7剂量水平(100 μg/kg)起观察到客观缓解;6例可评估的患者中有1例达到CR以及2例达到PR,ORR为50.0%。
在实体瘤中,从第7剂量水平(100 μg/kg)起也开始观察到多例PR和疾病稳定(SD)伴肿瘤负荷减小,包括非小细胞肺癌(NSCLC;1例PR及3例SD),胰腺癌(1例PR),三阴性乳腺癌(TNBC;1例SD),卵巢癌(1例SD);基于以上疗效趋势,随着剂量的增加,在实体瘤患者中有望观察到更好的疗效。