模仿HR2的磺酰基-γ-AA肽(环拟肽)是一种强效泛冠状病毒融合抑制剂,具有高度血脑屏障穿透性、长半衰期及良好的口服生物利用度
An HR2-Mimicking Sulfonyl-γ-AApeptide Is a Potent Pancoronavirus Fusion Inhibitor with Strong Blood−Brain Barrier
Permeability, Long Half-Life, and Promising Oral Bioavailability
Songyi Xue, Wei Xu*, Lei Wang, Xinling Wang, Qianyu Duan, Laurent Calcul, Shaohui Wang, Wenqi Liu, Xingmin Sun, Lu Lu*陆路 复旦大学病原微生物所, Shibo Jiang*姜世勃 复旦大学病原微生物所, and Jianfeng Cai 蔡健峰 南佛罗里达大学化学系*
pubs.acs.org/doi/10.1021/acscentsci.3c00313
文章概述:
一篇发表于2023年4月的《ACS Central Science》杂志上的研究揭示了一种新型的模仿HR2(heptad repeat 2)结构的磺酰基-γ-AA肽(环拟肽),这是一种针对冠状病毒的强大融合抑制剂。这项研究由多个机构的科研人员合作完成,包括复旦大学、南佛罗里达大学等,(据此也有研制了艾滋病的膜融合抑制剂)
研究亮点:
泛冠状病毒抑制作用:该肽模拟了SARS-CoV-2 S2亚单位中HR2的关键残基,并与HR1相互作用,有效阻止了病毒与细胞膜之间的融合,对SARS-CoV-2和其他人类冠状病毒展现出广泛的抑制活性。
血脑屏障穿透性:这种肽表现出强大的血脑屏障穿透能力,意味着其可能在治疗神经系统受累的冠状病毒感染中发挥作用。
长半衰期和口服生物利用度:与许多传统肽类药物相比,该肽具有更长的体内半衰期和有希望的口服生物利用度,这解决了传统肽类药物需要注射给药且易被酶降解的局限性。
设计与应用:研究团队开发了一系列螺旋状肽拟物(peptidomimetics),即D-磺酰基-γ-AA肽(环拟肽),这些分子能够精确模拟关键蛋白结构域,为设计抗病毒药物提供了新策略。
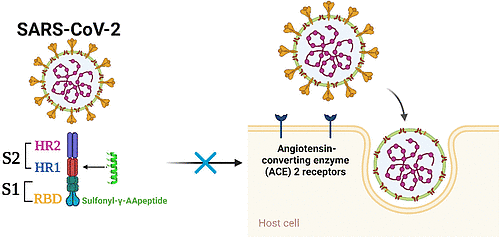
介绍
由SARS-CoV-2引起的COVID-19,截至2023年1月7日,已关联超过6.63亿确诊病例和670万死亡病例。【1】尽管目前已有多款疫苗【2】和小分子药物【3】获得授权或批准用于人类,但新病毒变异株的不断出现迅速威胁到了这些疫苗和药物的有效性。【4】因此,继续开发替代性和广谱的预防及治疗手段以对抗这一大流行病显得至关重要。
SARS-CoV-2属于高突变性的β-冠状病毒群体,这使得它们能够适应新的宿主和生态位。5其病毒颗粒由四种结构蛋白组成:刺突(S)、包膜(E)、膜(M)和核(N)蛋白。【6】这些蛋白在病毒生命周期中发挥关键作用,并且是所有人类冠状病毒(HCoVs)所共有的,这使它们成为开发广谱抗病毒药物的潜在目标。【7】其中,S蛋白通过招募细胞丝氨酸蛋白酶TMPRSS2对S蛋白进行预处理以及利用血管紧张素转换酶2(ACE2)作为入侵受体,促进病毒进入靶细胞(图1A)。【8,9】S蛋白包含两个功能亚单位域(图1A,B),即S1和S2。10S1亚单位通过其受体结合域(RBD)与ACE2受体结合,随后S2亚单位发生构象变化,允许融合肽域(FP)插入到宿主靶细胞的细胞膜中。S2亚单位中的七重重复区1(HR1)形成同源三聚体结构,暴露出三个高度保守的疏水沟槽,与七重重复区2(HR2)相互作用(图1C,D),形成六螺旋束(6-HB)结构,使病毒膜与细胞膜紧密接触,启动病毒融合过程(图1B)。【7,11】RBD和HR区域是针对融合过程开发特定治疗药物的极好靶点。12尽管基于RBD的抗体已被证实能有效阻止病毒附着于ACE2,【13】但由于RBD的高度可变性,设计针对RBD的广谱抗病毒药物极具挑战性。【14,15】相比之下,普遍认为HR1可以作为一个良好的靶点来开发针对高度致病性HCoVs的泛冠状病毒融合抑制剂,因为其在不同HCoVs中高度保守。【7,16】
图1. SARS-CoV-2感染的融合过程及磺酰基-γ-AA肽(环拟肽)抑制机制的解析。
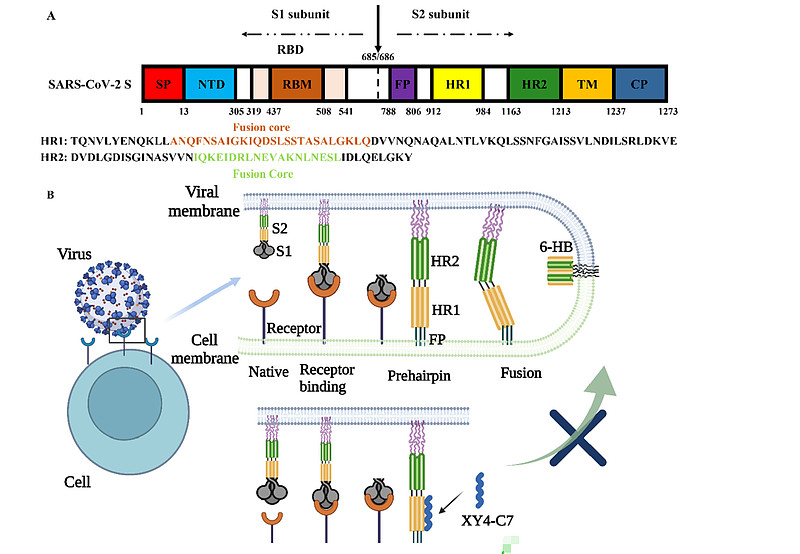
A) SARS-CoV-2刺突蛋白的示意图。SP表示信号肽;NTD表示氨基端结构域;RBD表示受体结合域;RBM表示受体结合基序;FP表示融合肽;HR1表示七肽重复序列1;HR2表示七肽重复序列2;TM表示跨膜域;CP表示胞质域。
(B) 由SARS-CoV-2的S蛋白介导的融合过程,以及磺酰基-γ-AA肽(环拟肽)提出的抑制SARS-CoV-2感染的机制。
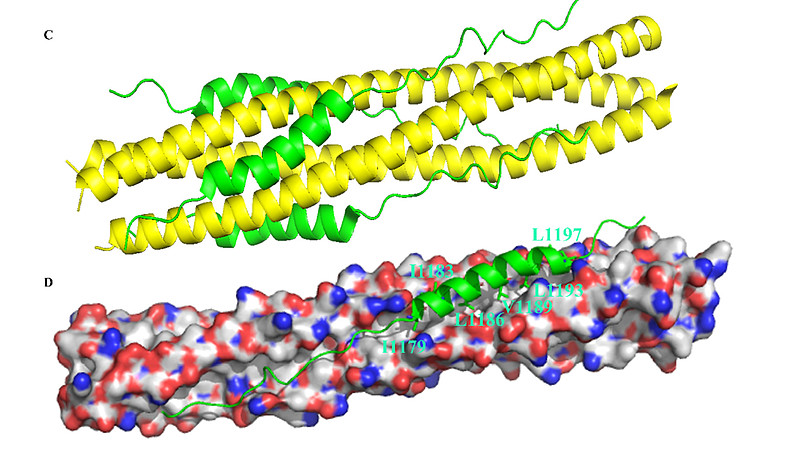
(C) HR2与HR1相互作用形成6螺旋束(6-HB)的晶体结构侧视图(PDB代码:6LXT)。
(D) HR2上关键残基(绿色)与HR1三聚体上的口袋之间的结合互动。
已有一些针对HR1的肽类融合抑制剂被研发出来,并显示出在体外和体内显著抑制SARS-CoV-2感染的效果。【7,16-21】姜及其同事鉴定了两种泛冠状病毒融合抑制剂EK1【7】和EK1C4,【16】它们能显著抑制包括SARS-CoV-2、SARS-CoV、MERS-CoV、HCoV-NL63和HCoV-OC43在内的多种人冠状病毒的融合。朱等人开发了一种脂肽融合抑制剂IPB02,【17】该抑制剂在防止SARS-CoV-2 S蛋白介导的细胞-细胞融合和假病毒感染方面表现出高效。德弗里斯等开发的二聚脂肽融合抑制剂[SARSHRC-PEG4]2-chol对SARS-CoV-2感染的雪貂进行每日鼻内给药,能完全阻止SARS-CoV-2的直接接触传播。【18】然而,这些生物活性肽由于其典型的肽主链结构,固有地具有低生物稳定性和生物可利用性,导致半衰期短,难以作为口服药物使用。【22】
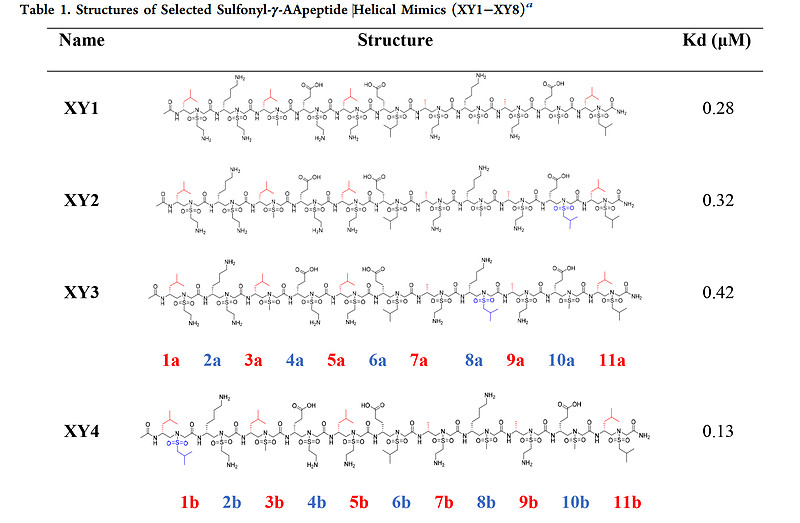
非天然序列特异性肽模拟物已成为调节蛋白质-蛋白质相互作用(PPIs)以缓解肽固有缺点问题的一个有前景的替代策略。【23-27】除了保留天然肽的优点外,折叠肽模拟物还展现出独特的结构和功能,并且对酶水解具有高度抵抗性。【23,28】我们基于γ-手性肽核酸(PNA)主链开发出一类新的肽模拟物——γ-AA肽(环拟肽)【28-30】(N-酰基-N-氨基乙基氨基酸的寡聚物),它们展现出对蛋白水解降解的非凡抵抗性和化学多样化的可能性,使之成为多种生物学应用的理想候选者。【12,31-34】作为γ-AA肽(环拟肽)的一个子类,磺酰基-γ-AA肽(环拟肽)(图2A)不仅具有上述优点,还能采取明确界定的螺旋结构【33-36】(图2B,C)。值得注意的是,磺酰基-γ-AA肽(环拟肽)螺旋展现出比相同长度α-螺旋更强大和稳定的螺旋构象,这可能归因于分子框架内磺酰胺基团的分子内氢键和弯曲性质。众所周知,均一的L-磺酰基-γ-AA肽(环拟肽)采取左手414-螺旋构象,每圈四个侧链,螺旋螺距为5.1 Å35(图2B,C)。由于这些特性与α-螺旋相似,只是手性相反,因此它们被设计用来模拟蛋白质的螺旋域,并已成功调控了包括BCL9、37p53、38GLP-1、39和VEGF在内的多个PPIs。【40,41】值得注意的是,序列中的左手性可以通过将L-磺酰基-γ-AA构建块改为D-磺酰基-γ-AA构建块(图2D)切换到右手性,导致的右手螺旋由于与α-螺旋更接近而预期能进一步促进α-螺旋模拟物的设计(图2E,F)。【36,42】我们设想,通过合理设计,D-磺酰基-γ-AA肽(环拟肽)的分子骨架可用于抑制SARS-CoV-2的病毒融合过程。
图2. 磺酰基-γ-AA肽(环拟肽)的化学结构与晶体结构。
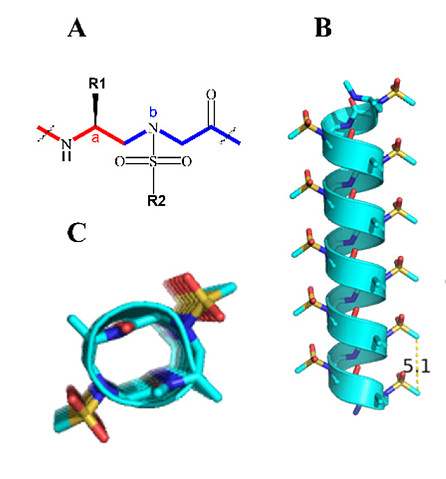
(A) L-磺酰基-γ-AA肽(环拟肽)的化学结构。a和b分别代表来自构建单元的手性侧链和磺酰胺侧链。
(B) L-磺酰基-γ-AA肽(环拟肽)的晶体结构。 (C) (B)的顶视图。
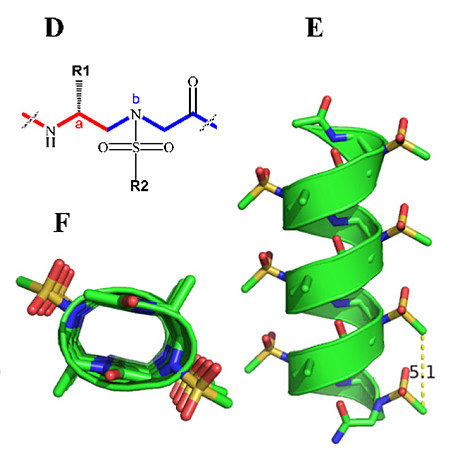
(D) D-磺酰基-γ-AA肽(环拟肽)的化学结构。a和b同样分别指构建单元的手性侧链和磺酰胺侧链。
(E) D-磺酰基-γ-AA肽(环拟肽)的晶体结构。
(F) (E)的顶视图。
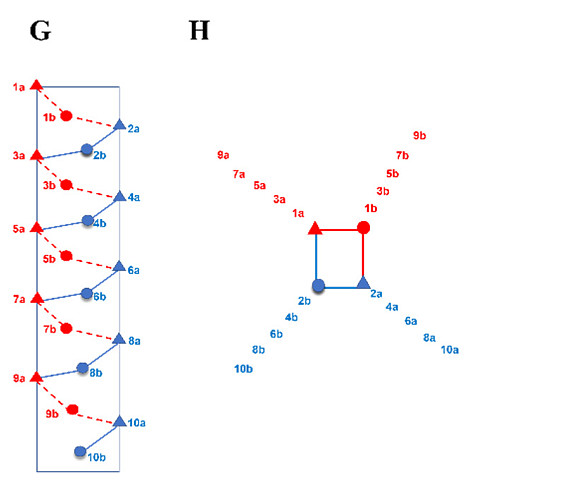
(G, H) 磺酰基-γ-AA肽(环拟肽)侧链分布的示意图,从(G)侧面视图和(H)螺旋轮的顶视图展示。
据我们所知,目前尚无口服生物可用的融合抑制肽,也无基于完全非天然折叠支架能阻断SARS-CoV-2进入的融合抑制剂。本文报道了设计出模仿HR2肽热点并破坏SARS-CoV-2 S2亚单位中HR1和HR2域之间相互作用的右手螺旋D-磺酰基-γ-AA肽(环拟肽)。通过将胆固醇分子偶联到领先的磺酰基-γ-AA肽(环拟肽)上,我们鉴定出一种脂磺酰基-γ-AA肽(环拟肽)(XY4-C7),它在假病毒(PsV)和真实病毒感染试验中表现出强烈的抑制活性,呈现出高选择性指数(SI)。此外,XY4-C7展示了对抗一系列冠状病毒的广谱抗病毒活性,包括SARS-CoV-2及其Delta变异株以及其他HCoVs如SARS-CoV、MERS-CoV、HCoV-NL63、HCoV-OC43以及蝙蝠SARS相关冠状病毒(SARSr-CoV)WIV1,这与HR2和HR1域在不同冠状病毒中高度保守的事实相符。此外,鼻内途径给予XY4-C7的体内研究表明,其具有高度的预防效果和优异的治疗效果。此外,由于其非天然的主链结构,XY4-C7表现出对蛋白水解降解的显著抵抗性,在药代动力学(PK)研究中展现出非常长的半衰期和有希望的口服生物利用度,表明磺酰基-γ-AA肽(环拟肽)有潜力开发成治疗和预防SARS-CoV-2和其他HCoVs感染的药物。
结果
六螺旋束形成的结构洞见:HR1与HR2间的关系
SARS-CoV-2 S蛋白介导的膜融合过程中,HR1与HR2域形成的六螺旋束(6-HB)至关重要,其晶体结构近期已被解析。【16】如上面的图1C、D所示,三条HR1分子构成一个平行的三螺旋卷曲核心,周围由三条反平行的HR2螺旋包围。疏水力驱动HR1与HR2域之间的相互作用,这一作用主要位于螺旋融合核心区域。每一对相邻的HR1螺旋形成一个显著的疏水沟槽,作为HR2域中I1179、I1183、L1186、V1189、L1193和L1197等疏水残基的结合位点。所有这些疏水残基都位于HR2 α-螺旋的同一面上(图1D)。此外,侧链间的亲水性相互作用也有助于稳定束结构。
基于理性设计的磺酰基-γ-AA肽(环拟肽)模拟融合核心中的HR2肽
基于以上分析,我们在D-磺酰基-γ-AA肽(环拟肽)螺旋的同一面上1a、3a、5a、7a、9a和11a位置引入六个手性疏水残基,分别模拟HR2的I1179、I1183、L1186、V1189、L1193和L1197与HR1的结合相互作用,以复现与HR1的结合亲和力(上面图2D、H)。为了增强序列的螺旋稳定性和溶解性,在另外两个面上引入了负电和正电荷侧链形成盐桥(表1)。我们首先设计并合成了四个序列(XY1至XY4,表1),在1b、8b和10b位置带有不同大小的疏水基团,因为这些残基位于HR1的疏水沟槽中。我们首先通过荧光偏振实验评估这些序列与HR1肽(911-987)的结合亲和力。不出所料,所有四个序列都显示了与HR1肽的极佳结合亲和力,Kd值在0.13至0.42 μM之间(表1)。这与模型预测一致,其中HR2的关键残基(图3A)与XY4的疏水侧链(图3B)完美重叠(图3C)。XY4与HR2肽在HR1表面的叠加(图3E)也提示XY4能有效地识别HR1的疏水裂隙。实际上,所有D-磺酰基-γ-AA肽(环拟肽)在溶液中均呈现典型的右手螺旋结构。如图3F所示,圆二色(CD)实验揭示了在205至215 nm间强烈的负Cotton效应,42这是左旋L-磺酰基-γ-AA肽(环拟肽)CD特征的镜像,意味着这些D-磺酰基-γ-AA肽(环拟肽)采取了类似于α-螺旋肽的右手螺旋构象。
图3. 基于磺酰基-γ-AA肽的进入抑制剂的合理设计及其体外抑制活性评估。
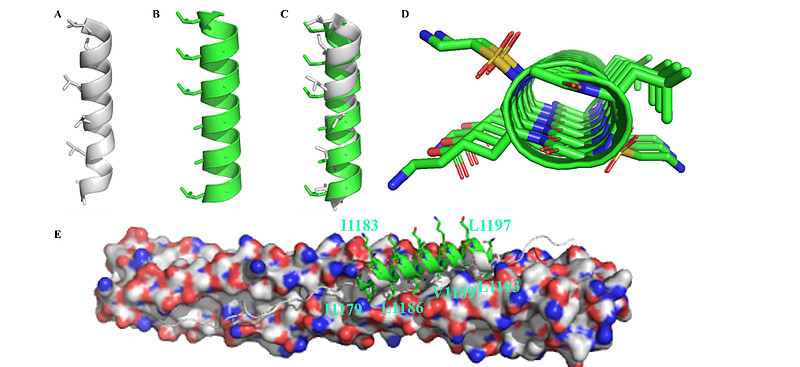
(A) 融合核心中HR2肽的结构(白色)。
(B) 作为模拟物的磺酰基-γ-AA肽XY4(绿色)。
(C) (A)和(B)间关键结合残基的重叠。
(D) (B)的顶视图,展示了极性侧链和关键疏水残基。
(E) XY4(绿色)与融合核心中HR2肽的关键残基在HR1结合表面上的叠加。
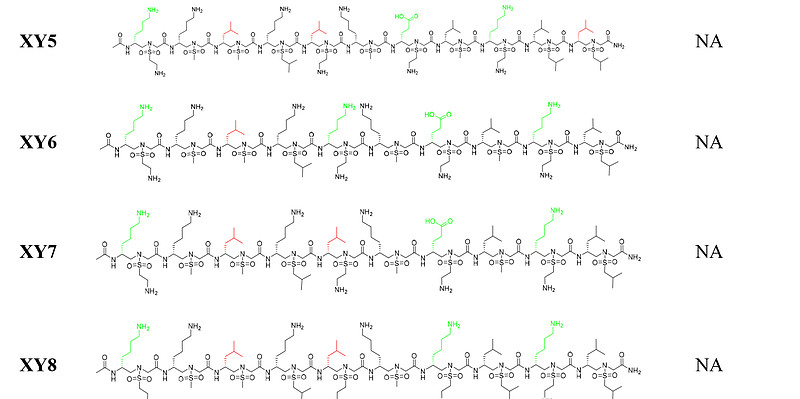
关键结合残基以红色显示。序列与HR1肽的结合亲和力(Kd)通过荧光偏振法确定。融合核心中HR2肽的氨基酸序列为SVVNIQKEIDRLNEVAKNLNESLIDLQ。
接下来,我们使用在Caco-2细胞中建立的SARS-CoV-2假病毒(PsV)感染实验,在20 μM浓度下测试了这四个序列的抑制活性。虽然效力不是特别高,但它们都展现了一定程度的抑制活性,特别是化合物XY4,在此浓度下抑制PsV感染约40%(图3G)。我们还合成了XY5、XY6、XY7和XY8,其中某些位于1a、3a、5a、7a、9a和11a位置的疏水残基被替换为亲水基团(表1)。正如预期,它们与HR1肽的结合亲和力非常低,确认了强有力的结合亲和力来源于磺酰基-γ-AA肽(环拟肽)成功模拟了融合核心中HR2肽的关键疏水残基。鉴于XY4表现出了最有效的抑制活性,因此被选为我们进一步改造的领头化合物。
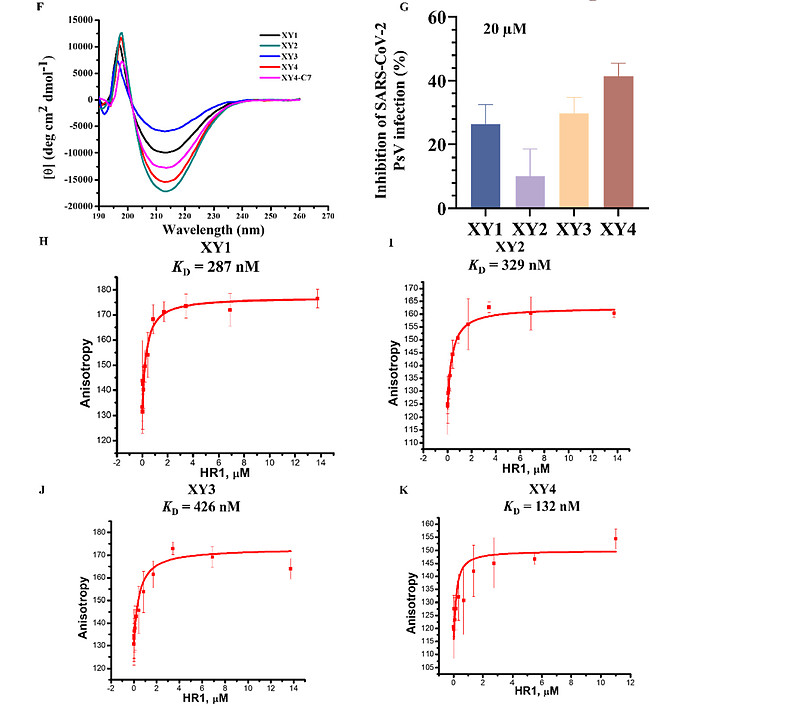
图3. 基于磺酰基-γ-AA肽的进入抑制剂的合理设计及其体外抑制活性评估。
(F) 磺酰基-γ-AA肽XY1-XY4及XY4-C7在PBS缓冲液中、室温下、100 μM浓度下的圆二色谱(CD)图谱。
(G) 四个领先化合物(XY1、XY2、XY3和XY4)在20 μM浓度下,通过SARS-CoV-2假病毒感染试验测定的抑制活性。
(H-K) HR1肽与(H) XY1、(I) XY2、(J) XY3及(K) XY4之间结合亲和力的测定,采用荧光偏振法。
XY4-C7,一种磺酰基-γ-AA肽(环拟肽)-PEGn-胆固醇缀合物,展现出了卓越的融合抑制活性和适度的细胞毒性。
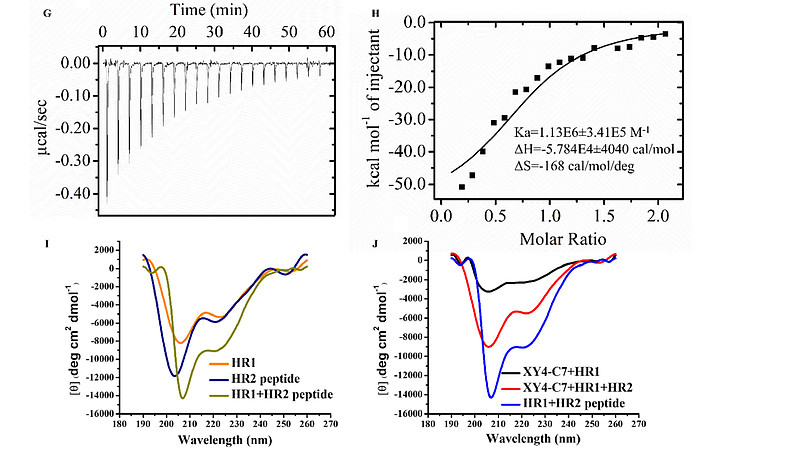
尽管XY4能够抑制SARS-CoV-2感染,但其活性明显弱于最近报道的泛冠状病毒融合抑制剂EK1。【7】脂质化是一种已验证的策略,通过增加融合抑制剂(如EK1C4)在宿主细胞膜表面的局部浓度来增强其抗病毒活性。【16,43】因此,借助不同长度的柔性连接子,如Fmoc-6-氨基己酸(6-Ahx)和胆固醇-聚乙二醇(PEG),将胆固醇共价连接到XY4的C末端,构建了相应的XY4-PEGn-胆固醇(图4A)。这些XY4-PEGn-胆固醇通过SARS-CoV-2假病毒(PsV)感染试验,以其半数最大抑制浓度(IC50)进行了评估。首先,我们添加了两个柔性连接子6-Ahx和不同长度(4、8和12个单元)的Chol-PEG。这三个化合物的IC50值分别为29.82、2.86和0.93μM(图4B),表明脂质化修饰成功,且PEG单元增多增强了抑制活性。我们用γSγS(图4A)替换了6-Ahx柔性连接子,灵感来自于EK1C4使用的刚性连接子(GSGSG),【16】从而得到了其他四个序列XY4-C4、XY4-C5、XY4-C6和XY4-C7。在Caco-2细胞中检测了这四种化合物的抗PsV活性,它们的IC50值分别为35.77、4.05、2.63和0.79μM(图4B),其中XY4-C7的活性远强于XY4,甚至优于之前报道的EK1肽(SLDQINVTFLDLEYEMKKLEEAIKKLEESYIDLKEL,IC50=2.38μM)。【7,16】因此,我们选择XY4-C7作为领先化合物进行进一步评估。值得注意的是,XY4-C7在剂量依赖性方式下有效地抑制了Caco-2细胞中SARS-CoV-2的真实感染,其IC50为0.24μM(图4C),与PsV感染试验结果一致。使用相同的细胞系评估其细胞毒性,半数最大细胞毒性浓度(CC50)为14.89μM(图4E)。选择性指数(SI=CC50/IC50)为62.04,表明XY4-C7特异性抑制SARS-CoV-2进入宿主细胞。后续实验也证明XY4-C7以剂量依赖的方式靶向S蛋白,抑制了SARS-CoV-2 S蛋白介导的细胞-细胞融合(图4D)。通过荧光偏振(FP)和等温滴定量热法(ITC)测定,确定了XY4-C7对HR1肽的结合亲和力,IC50值分别为0.1和0.8μM,表明向XY4添加胆固醇并未显著改变其对HR1肽的结合活性(图4F-H)。随后,我们采用圆二色光谱(CD)探查了XY4-C7抑制活性的机制。HR1肽和HR2肽在溶液中均表现出典型的α螺旋特性;HR1肽与HR2肽混合后,α螺旋特性更为显著(图4I),可能表明HR1/HR2复合物的形成。然而,在加入XY4-C7的情况下,HR1肽的CD特征强度显著下降,暗示由于HR1肽与XY4-C7的相互作用导致了显著的构象变化。此外,当XY4-C7与HR1肽/HR2肽共同混合时,6-HB的α-螺旋特性显著降低(图4J),表明XY4-C7通过强烈结合HR1,能够破坏HR1/HR2复合物的形成(图4F-J)。综上所述,这些结果表明XY4-C7是一种针对SARS-CoV-2感染的高效、选择性抑制剂,对HR1肽具有高结合亲和力,能够破坏HR1和HR2融合核心之间6-HB的形成。
图4. 磺酰基-γ-AA肽-PEGn-胆固醇抑制剂的合理设计及其体内的抗病毒活性评估。
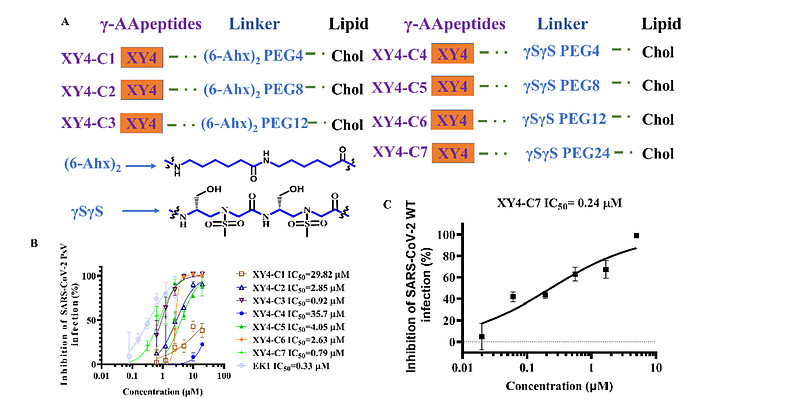
(A) 磺酰基-γ-AA肽-PEGn-胆固醇的设计图,包括XY4-C1至XY4-C7。
(B) 磺酰基-γ-AA肽-PEGn-胆固醇对SARS-CoV-2假病毒(PsV)感染的抑制活性。
(C) XY4-C7对Caco-2细胞中真实SARS-CoV-2感染的抑制活性。
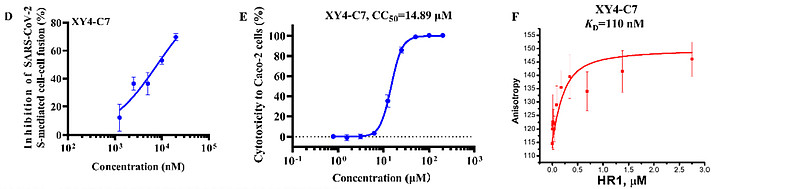
(D) XY4-C7对SARS-CoV-2 S蛋白介导的细胞-细胞融合的抑制活性。
(E) XY4-C7对Caco-2细胞的细胞毒性。
(F) 通过荧光偏振法测定XY4-C7与HR1肽之间的结合亲和力。
(G, H) 通过等温滴定热量法(ITC)测定XY4-C7与HR1肽之间的结合亲和力。
(I) SARS-CoV-2 HR1肽单独(橙色)、SARS-CoV-2 HR2肽(深蓝色)及SARS-CoV-2 HR1/HR2肽复合物(暗黄色)的圆二色谱(CD)图谱。
(J) XY4-C7/HR1肽混合物(黑色)、XY4-C7/HR1肽/HR2肽混合物(红色)及SARS-CoV-2 HR1/HR2肽复合物(蓝色)的CD图谱。
XY4-C7高效抑制了SARS-CoV-2 Delta变异株、三种假型HCoVs以及一种假型蝙蝠SARSr-CoV的感染。
为了确定XY4-C7的作用范围,我们测试了其对SARS-CoV-2 Delta变异株、另外三种HCoVs和一种蝙蝠SARSr-CoV的抑制活性。我们发现,XY4-C7在Vero-E6细胞中对SARS-CoV-2 Delta变异株的真实感染具有抑制作用,IC50值为4.73μM(图5A)。XY4-C7还能在不同的细胞系中强有力地抑制SARS-CoV、MERS-CoV、HCoV-NL63以及蝙蝠SARSr-CoV WIV1的假病毒感染,IC50值在0.81至9.42μM之间,证实了XY4-C7是一种泛HCoV融合抑制剂(图5B-E)。总的来说,XY4-C7是对SARS-CoV-2及其Delta变异株,以及其他HCoVs和可能导致未来冠状病毒疾病的蝙蝠SARSr-CoV都有效的有前景的广谱抗病毒剂。
图5. XY4-C7对真实SARS-CoV-2 Delta变异株、三种假型人冠状病毒、一种蝙蝠SARS相关冠状病毒的体外抑制作用,以及对小鼠体内人冠状病毒OC43感染的抑制作用。
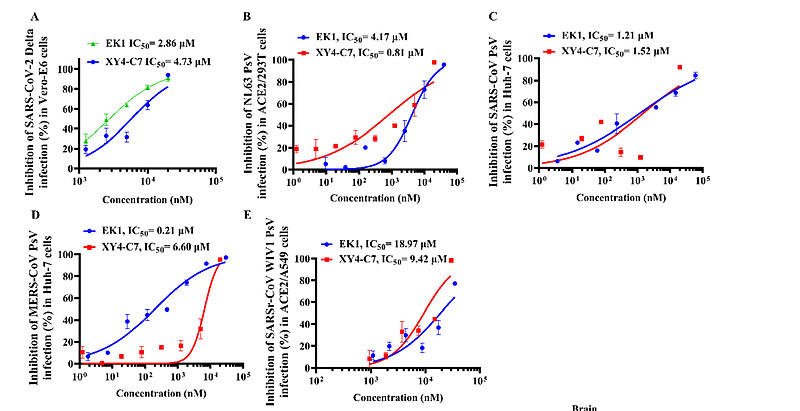
(A) XY4-C7对VeroE6细胞中真实SARS-CoV-2 Delta变异株感染的抑制活性。
(B) XY4-C7对ACE2/293T细胞中假型HCoV-NL63感染的抑制活性。
(C) XY4-C7对Huh-7细胞中假型SARS-CoV感染的抑制活性。
(D) XY4-C7对Huh-7细胞中假型MERS-CoV感染的抑制活性。
(E) XY4-C7对ACE2/A549细胞中假型蝙蝠SARSr-CoV W1V1感染的抑制活性。
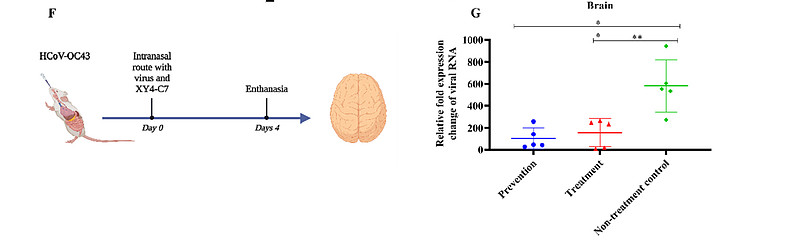
(F) XY4-C7施用与HCoV-OC43攻毒试验的示意图。
(G) XY4-C7(1 mg/kg)对新生小鼠HCoV-OC43感染的体内疗效。检测各组小鼠感染后第四天脑组织中的病毒RNA表达水平。
鼻内施用XY4-C7有效保护新生小鼠免受HCoV-OC43感染。
接下来,我们利用HCoV-OC43感染的小鼠模型,调查了XY4-C7在临床应用中的保护效果。在预防组(n=5)或治疗组(n=5)中,通过鼻内途径,以低单剂量1mg/kg,在HCoV-OC43 100 TCID50暴露前0.5小时或后,对OC43感染的新生小鼠施用XY4-C7。小鼠在4天后被解剖,摘取大脑以测定病毒载量。如图5G所示,预防组和治疗组的HCoV-OC43 RNA水平均显著低于未治疗组。这一结果表明,XY4-C7能有效保护新生小鼠免受HCoV-OC43感染,基于此证据,可以合理推测XY4-C7也能有效抑制体内SARS-CoV-2及其他HCoVs的感染。
XY4-C7在胰蛋白酶或人血清中展现出高度稳定性。
对于传统的肽类化合物而言,它们在面对蛋白酶降解时固有的敏感性是开发抗病毒药物的一大瓶颈。因此,我们评估了在胰蛋白酶(这是一种理论上能将肽分解为单一氨基酸的混合水解酶)或人血清环境中XY4-C7与HR2肽的稳定性。样本在胰蛋白酶或人血清中孵育24小时后,通过液相色谱-串联质谱(LC/MS/MS)进行分析(图6A)。结果表明,XY4-C7具有高度稳定性,在24小时内未观察到显著降解;而HR2肽在胰蛋白酶中被完全降解,在人血清中也有约90%被降解。
图6. 在小鼠模型中评估XY4-C7的稳定性、膜被动渗透性、口服生物利用度和药代动力学概况。
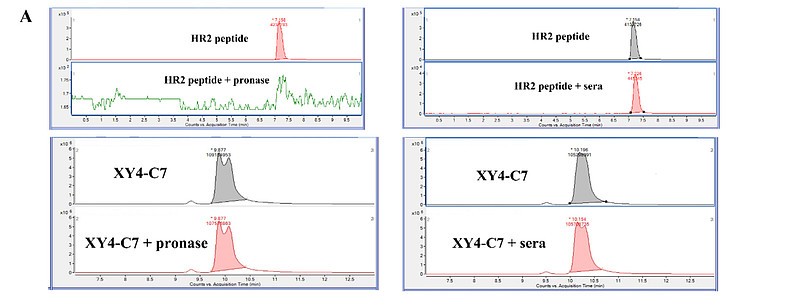
(A) HR2肽模拟物稳定性研究。分别在含有胰蛋白酶或人血清的条件下,对指定对照和XY4-C7进行24小时孵育后的LC/MS/MS分析。
XY4-C7对血脑屏障及胃肠屏障膜显示出良好的被动渗透性。
平行人工膜渗透性试验(PAMPA)是一种广泛应用于制药行业以评估药物候选物渗透性的高通量渗透性测试。44基于此,我们利用PAMPA评估了XY4-C7通过血脑屏障(BBB)的渗透性(PAMPA-BBB)和通过胃肠道(GIT)的渗透性(PAMPA-GIT)。在这两种测试中,预期的渗透系数(Papp)值,对于良好、中等和低渗透性分别是>20×10^-6 cm/s、1-20×10^-6 cm/s和<1×10^-6 cm/s。考虑到人体内腔pH值在胃、十二指肠、回肠、盲肠和结肠中有所变化,我们有必要在不同pH条件下评估XY4-C7对PAMPA-GIT的渗透能力。我们使用了全口服生物可利用药物卡马西平(50 μM)作为阳性对照,它在三种不同pH值下具有约150×10^-6 cm/s的较高Papp值;而口服生物可利用性差的药物安替比林(200 μM)则作为阴性对照,其在相同pH值下的Papp值<6×10^-6 cm/s(图6B)。如图6B所示,XY4-C7(50 μM)在pH 5.0、6.2和7.4下分别表现出有利的渗透性,Papp值分别为372×10^-6 cm/s、344×10^-6 cm/s和306×10^-6 cm/s,显著高于标准值。因此,预期XY4-C7在体内具有非常可观的口服生物可利用性。
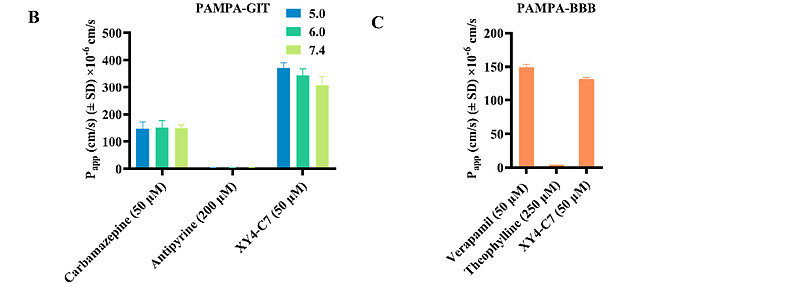
(B) 不同pH条件下标准物质和XY4-C7的PAMPA-GIT(胃肠道透膜性平行人工膜渗透性试验)。该图展示了不同pH值(5.0、6.2和7.4)下,阳性对照药物卡马西平、阴性对照药物安替比林与XY4-C7的渗透率(Papp值),显示XY4-C7在各种胃肠道相关pH值下具有高度的渗透性。
(C) pH 7.4下标准物质和XY4-C7的PAMPA-BBB(血脑屏障透膜性平行人工膜渗透性试验)。此图展示了在标准BBB条件下的渗透性评估,显示XY4-C7如同阳性对照药物维拉帕米一样,具有透过血脑屏障的能力,而阴性对照药物茶碱则不具备此能力。
SARS-CoV-2因其强大的入侵中枢神经系统(CNS)能力,并影响特定核团或神经回路的功能,导致包括急性中风、嗅觉减退、吉兰-巴雷综合症和脑炎在内的一系列严重神经症状和并发症。【45】因此,我们利用PAMPA-BBB试验评估了XY4-C7跨越血脑屏障(BBB)的潜力,预测其阻止SARS-CoV-2侵入CNS的能力,从而控制这些症状和并发症。阳性对照药物维拉帕米在50 μM浓度下能轻易穿过BBB,Papp值为148×10^-6 cm/s(图6C)。相比之下,作为阴性对照的茶碱即使在250 μM浓度下,Papp值也只有4×10^-6 cm/s,几乎不能穿过BBB(图6C)。与维拉帕米相似,XY4-C7(50 μM)能轻易穿透BBB,Papp值为132×10^-6 cm/s,这有助于解释为什么在HCoV-OC43感染小鼠模型中,它能显著保护小鼠大脑免受HCoV-OC43感染(图6C)。因此,我们可以预测XY4-C7在控制CNS中的SARS-CoV-2方面具有非常乐观的潜力。
XY4-C7在小鼠中的药代动力学特征显示出更长的半衰期和非常有希望的口服生物利用度。
为了确定XY4-C7的口服吸收和体内稳定性,我们通过腹腔注射(IP)和口服(OP)方式给予小鼠30 mg/kg的XY4-C7,并进行了48小时的药动学研究。在IP给药中,5小时内达到平均最大血液浓度(Cmax)408,769 μg/L;而在OP给药中,4小时后达到Cmax 114,140 μg/L(图6D、E)。这表明,两种给药方式的有效血液暴露量分别约为XY4-C7 IC50值的350倍和97倍。此外,两种给药方式的有效期均超过24小时,血浆浓度衰减的半衰期(t1/2)在IP给药中为9.4小时,在OP给药中显著延长至28.9小时(图6D、E)。最重要的是,XY4-C7再次显示出高达28%的口服生物利用度(F),表明它具有作为口服药物使用的极大潜力(图6D、E)。
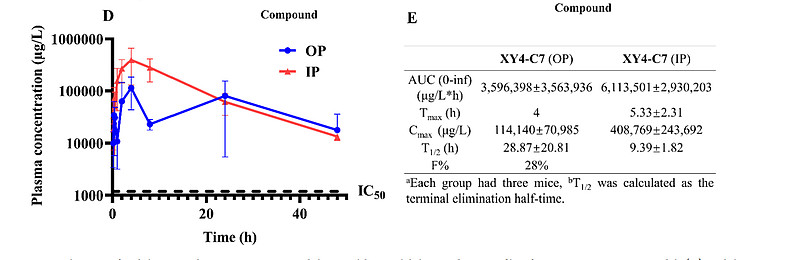
(D) XY4-C7药代动力学研究的时间-浓度曲线。红色线条代表腹腔注射(IP)给药30 mg/kg XY4-C7后,C57BL/6小鼠的血浆浓度随时间的变化;蓝色线条代表口服(OP)给药相同剂量后的血浆浓度变化。数据以平均值±标准偏差(SD)形式展现,n=3。虚线表示50%抑制率的阈值。
(E) 小鼠中XY4-C7在48小时内的药代动力学参数。该部分总结了XY4-C7在小鼠体内的药代动力学特性,包括半衰期(t1/2)、清除率(CL)、表观分布容积(Vd)、达峰时间(Tmax)、峰值浓度(Cmax)及口服生物利用度(F)等关键参数,强调了XY4-C7不仅具有良好的体内稳定性,而且口服后能快速达到有效血药浓度,显示出长期留存体内和高口服生物利用度的特性。
讨论
为了对抗SARS-CoV-2大流行以及未来可能出现的新兴和重新出现的人冠状病毒,特别是在全球未接种疫苗人群中以及对各种变异株产生药物抵抗性的日益担忧背景下,迫切需要开发具有广谱活性的长效口服药物。像EK1、EK1C4和[SARSHRC-PEG4]2-chol这样的生物活性肽,作为泛冠状病毒融合抑制剂,已经显示出对SARS-CoV-2的强大抑制活性。7,16,18然而,由于缺乏天然肽骨架,它们作为长效口服药物的使用面临生物利用度和生物稳定性有限的挑战。肽拟态体旨在模仿生物活性肽和蛋白质的结构与功能,已在蛋白质表面模拟和识别、调节蛋白质-蛋白质相互作用(PPIs)和催化等方面展现出显著的应用。最近,我们创建并应用了磺酰基-γ-AA肽(环拟肽)作为新螺旋框架来设计蛋白质螺旋结构域拟态体,以及调节多种与药物相关的PPIs,包括VEGF/VEGFR、p53/MDM2、GLP-1、BCL9/β-catenin等。这些大多由具有左手螺旋构象的L-磺酰基-AA肽(环拟肽)构建块构建,与α-肽的右手螺旋相反。本文中,我们报告了基于D-磺酰基-γ-AA肽(环拟肽)的右手螺旋折叠体,预期展示与α-螺旋更一致的右手螺旋构象,以模拟融合核心中的HR2肽,从而阻止SARS-CoV-2的融合过程。
当前设计基于HR2融合核心与HR1三聚体复合物的晶体结构。融合核心中HR2螺旋的关键残基,包括Ile1179、Ile1183、Leu1186、Val1189、Leu1193和Leu1197,参与了与HR1三聚体的结合。因此,设计了一些磺酰基-γ-AA肽(环拟肽),基于螺旋结构,在磺酰基-γ-AA肽(环拟肽)折叠体的同一面上分别在1a、3a、5a、7a、9a和11a位置引入手性侧链,以重现这些疏水功能。在我们折叠体的其他两个不参与与融合核心中HR1区相互作用的面上,加入了亲水性基团,以提高螺旋稳定性和溶解性。基于这些设计理念,我们展示了某些磺酰基-γ-AA肽(环拟肽)对HR1肽疏水表面具有优异的结合亲和力和强相互作用。这些结果证明了这些磺酰基-γ-AA肽(环拟肽)通过与HR1三聚体的相互作用成功模拟了融合核心中的HR2肽。
经过PsV感染试验验证,领先序列XY4被选作进一步优化。由于脂质化已知能提高融合抑制剂的效率,我们添加了两种不同的连接子(刚性和柔性),以及不同长度的PEG与Chol。我们发现XY4-C7保留了与HR1肽的结合亲和力和相互作用能力,并在真实的SARS-CoV-2感染试验中表现出极高的活性,IC50为0.24 μM,SI为62。此外,XY4-C7对SARS-CoV-2 Delta变异株、伪型SARS-CoV、MERS-CoV、HCoV-NL63以及蝙蝠SARSr-CoV WIV1也具有高度有效性。体外测试后,我们发现鼻内施用XY4-C7给HCoV-OC43感染的新生小鼠,有效抑制了其体内感染。最重要的是,体内外药代动力学研究表明,XY4-C7对蛋白酶降解具有高度抵抗力,半衰期极长,且具有非常有希望的口服生物利用度。
结论
我们已经鉴定了几种模拟SARS-CoV-2 S蛋白S2亚单位融合核心中HR2肽的非天然螺旋折叠体拟态体。经验证,我们发现领先化合物XY4-C7是对SARS-CoV-2及其Delta变异株、几种人冠状病毒(包括SARS-CoV、MERS-CoV、HCoV-NL63和HCoV-OC43)以及蝙蝠SARSr-CoV WIV1的高效泛冠状病毒融合抑制剂。此外,它在PAMPA和药动学测试中(包括口服和腹腔注射给药)显示出杰出的药代动力学性质。因此,有理由认为XY4-C7可以进一步开发成为一种新型口服抗人冠状病毒药物,与其它具有不同作用机制的可用COVID-19治疗药物联合使用,可能会产生协同抗病毒效应,形成针对SARS-CoV-2和其他人冠状病毒感染的新疗法。总体而言,我们认为这项工作不仅可扩展用于开发基于磺酰基-γ-AA肽(环拟肽)的不同抗病毒剂,也可用于调控成千上万的其他PPIs。