

5月7日,美国制药公司辉瑞在给PPMD(一家致力于解决杜氏肌肉营养不良症的社区性非营利机构)的信件中表示,参与2期“DAYLIGHT”研究的一名小男孩已经死亡。
辉瑞在信中透露,这名患有杜氏肌肉营养不良症的男孩于2023年年初接受了试验药物的治疗,死于心搏骤停。目前仍在进一步确认这一死亡事件是否与基因治疗的直接关联性。辉瑞表示,“我们尚未掌握完整信息,正在积极与试验现场调查人员合作,了解发生的情况。”


暂停III期临床
辉瑞名为DAYLIGHT的基因疗法临床试验,主要针对2岁或3岁的男性患儿,是评估辉瑞治疗杜氏肌肉营养不良症的候选药物——福达斯替尼莫瓦帕罗韦(fordadistrogene movaparvovec,研发代码:PF-06939926)的安全性和抗肌营养不良蛋白表达的试验。
据上述PPMD机构此前报道显示,于2022 年11月18日,辉瑞启动了2期DAYLIGHT研究中患者接受给药。DAYLIGHT临床试验有6个临床试验地点,设在了澳大利亚和美国,没有安慰剂组。当月早些时候,第一位患者在澳大利亚悉尼Westmead的儿童医院接受了给药。
在辉瑞的基因疗法中,还包括一项3期CIFFREO临床试验,主要针对4-8岁以内的患者男孩的安慰剂对照,以评估fordadistrogene movaparvovec的疗效与安全性。
按照辉瑞原本的计划,3期CIFFREO试验的初始给药也在2023年完成。如果结果积极,辉瑞将凭此申请上市。但由于患者死亡,目前辉瑞已经暂停了CIFFREO交叉部分的给药。
“除了暂停给药外,试验活动仍按计划进行,”辉瑞公司表示。
值得注意的是,这是辉瑞第二次宣布其DMD基因治疗试验中出现死亡事件。2021年9月,1名16岁患者在接受较高剂量治疗后死于心源性休克,美国食品药品监督管理局(FDA)随即暂停了辉瑞的DMD试验。
随后,辉瑞公司对试验方案进行修订,强化了安全性监控和药效测定标准,在2022年4月,该研究被批准重启进行。
有着175岁历史的辉瑞被誉为“宇宙大药厂”,除杜氏肌营养不良症(DMD)外,辉瑞目前还有另外两个基因治疗III期项目,分别是用于治疗中重度 B型血友病的BBEQVEZ和血友病A(giroctocogene fitelparvovec)。
今年4月26日,辉瑞公司递交的Beqvez新药上市申请已被FDA接受,定价为350万美元一针。

“传男不传女”的罕见病
杜氏肌肉营养不良症(Duchenne Muscular Dystrophy,DMD),是一种罕见的隐性遗传神经肌肉疾病,是所有肌营养不良中发病率最高、病情最为严重的一种类型。
DMD会导致肌肉萎缩、炎症和纤维化,并伴有进行性虚弱和呼吸衰竭。患者常见于在3至5岁起病,9至12岁丧失独立行走能力,多数患者因呼吸或心力衰竭在30岁前死亡。但由于系统护理的进步,许多年轻DMD患者的寿命和生存质量有着长足的改善。
DMD是由于患者编码抗肌萎缩蛋白的基因出现突变。女性通常为携带者,因为在另一条染色体上的正常基因仍能发挥作用,所以女性往往不发病,或症状很轻。该罕见病也被俗称为“传男不传女”。
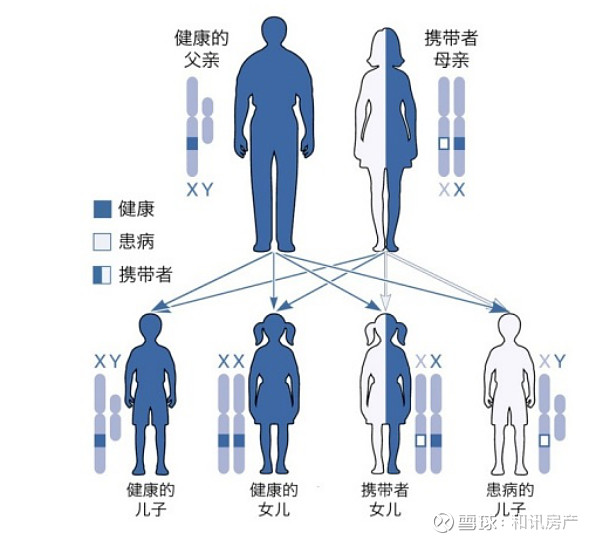
据许多专家医生科普表示,作为最常见的罕见遗传病之一,DMD的发病率是大约每3500-5000名男性新生儿中就有1名患儿。其中,80%为遗传性,20%为感染、过敏、退行性或增生性疾病。
在我国,每年约有400-500例DMD患儿出生,累计约7万人确诊为DMD,是世界上该病患者人数最多的国家之一。
虽然DMD罕见病一直是全球药企研发的“热门”,且有多款药品的临床试验在全球展开,但目前,杜氏肌营养不良症仍无法治愈。全世界患者的一线基础治疗都是以激素治疗和康复锻炼为主,以控制症状和减缓疾病进展为目的。
糖皮质激素治疗是DMD的经典疗法,常见用药包括泼尼松、甲泼尼龙片和地夫可特。但据很多患者家庭表示,激素药物大多存在较大的副作用。比如,可能会降低患者的免疫能力,患者易发感染,也可能会造成骨质疏松、消化性溃疡、高血压、糖尿病、肥胖等多种症状。
而且,即使是采取激素和保健药品治疗,一年也需要花费4万元到5万元。

一针310万美元的疗法
近年来,DMD的新疗法更多是从基因缺陷病因入手。如果辉瑞的fordadistrogene movaparvovec能最终获批上市,它将成为DMD患者可用的第2个基因疗法。
第一个是美国生物制药公司Sarepta和罗氏制药联合开发的Elevidys。2023年6月,Elevidys获得美国FDA批准上市。Elevidys是一种基于腺病毒(AAV rh74)的重组基因疗法,据Sarepta称,该疗法可以对所有类型的DMD患者有效,且仅需单次静脉输注给药。
但值得注意的是,Elevidys获得FDA的加速批准和成功上市,让许多人感到惊讶并引起一些业内人士的反对。
其争议点在于,Elevidys在3期临床试验中仅到达了次要终点,未能达到主要终点。也即Elevidys似乎对患者的肌肉功能只有适度的影响,而且只对某些DMD患者有影响;并且FDA在批准Elevidys的同时,限定了其适用对象为4至5岁的儿童,而不是Sarepta临床试验中的4至7岁受试者群体。
更大的争议在于,Sarepta将Elevidys的价格设定为每剂310万美元,使其成为史上第二贵的基因治疗产品。现阶段,最贵的基因疗法是去年11月获批的用于血友病治疗的产品Hemgenix,定价为350万美元。
“对于一种在2项随机试验中未能达到主要终点的疗法来说,这是一个巨大的代价,而且显然不是治愈性的。”有从事临床与经济审查药物定价研究的业内人士如此评价。
但面对“求药若渴”的罕见病群体,不少家庭还是选择为其高昂药价买单。
2023年8月,Elevidys进行正式销售交付,而据公司2023财报显示,Elevidys销售额达2.004 亿美元,销售额远超预期;尤其是在2023年第四季度,Elevidys的销售额达到1.313亿美元。
和讯网搜索国家药监局官网发现,在中国,目前尚无治疗杜氏肌营养不良症的治疗药物获批。但值得期待的是,一款温和激素药物在中国的上市或许指日可待。
今年2月18日,中国国家药监局药品审评中心(CDE)官网公示,上海曙方医药申报的vamorolone口服混悬液拟纳入优先审评,针对的适应症为4岁及以上杜氏肌营养不良(DMD)患者。
温和激素Vamorolone是曙方医药花费1.24亿美元自瑞士制药公司Santhera Pharmaceuticals引进的一款罕见病新药,目前该药在欧美、美国和德国均已上市。
据一名DMD患者家属分享,温和激素Vamorolone相比传统激素,可以大大减少家长们重点关注的一些副作用。它的特点是可在保留传统激素抗炎效果的同时,减轻部分由传统激素带来的副作用,以此延长DMD 患者的激素使用时间,进一步减缓病程。
“当然,这款药物属于罕见病药物,从目前的定价来看,对于国内DMD患者来说是偏高的,将是地夫可特价格的几十倍。”该患者家属表示。
据了解,今年5月13日起,可以在北京协和医院购买的西班牙版地夫可特的价格为70元。
“对于DMD患者而言,激素使用并不是一次性的,有可能是一辈子的。”患者家属表示,大多医生建议DMD患者三岁半开始使用激素,按目前DMD患者平均寿命30岁来说,一个DMD患者如果进行激素规范治疗的话,使用激素的时间可能长达20多年,除开别的药物、保健品或者辅助设备,光是激素用药就将是一笔上百万的花费。
他认为,如果这款药物不能实现医保报销的话,国内的DMD家庭将面临着两难选择。
--THE END--
