
6月17日,凭借mRNA新冠疫苗赚得盆满钵满的BioNTech发布公告表示,其ADC产品BNT326/YL202的临床被FDA叫停了,原因是FDA担忧较高剂量的YL202可能会使受试者面临不合理且显著的患病或伤害风险。
据公开信息,这是自2023年2月开始的多中心、开放标签、首次人体Ⅰ期临床,主要用于评估新药治疗晚期或转移性EGFR突变型非小细胞肺癌、HR+/HER2阴性乳腺癌患者的安全性和耐受性,以及在相关患者中的最大耐受剂量。
这款产品来源于一家中国医药界新星企业——宜联生物。上年10月,宜联生物将一款靶向HER3的ADC产品的中国区外的开发、制造和商业化独家权利,授予BioNTech。作为回报,宜联生物获得7000万美元首付款。
这款靶向HRE3的ADC产品,就是YL202。没想到,本是一桩新质生产力冒头的故事,却在8个月后被FDA打了个问号。
能让FDA对安全性感到担忧的,会是什么情况呢?答案呼之欲出,那就是入组患者出现了死亡样例。
6月初的ASCO大会上,宜联生物公布了这款产品Ⅰ期临床的数据。据其公布,该临床试验共分为7种不同剂量水平来进行,总体响应率(ORR)为42.3%,疾病控制率为94.2%,反应持续时间的中位数(DOR,N=52)达到5.8个月(95% CI;2.9,未达到),显示出较好的疗效持久性。
硬币另一面是,其中有三个死亡案例。其中两位患者在4.0mg/kg的剂量下,因发热性中性粒细胞减少症和败血症去世;一位患者在5.5mg/kg的剂量下,因感染了COVID-19并发展为间质性肺炎去世。
因感染新冠去世的还好跟监管说明情况。其余两例,就不是那么好办了。
通常来说,在临床试验中如果出现了较大的不良反应,PI们的第一反应,就是加快该分类下的患者入组,一方面是要从更多数据维度测试不良反应是否与产品相关,另一方面,也能先稳住这部分的不良反应率。
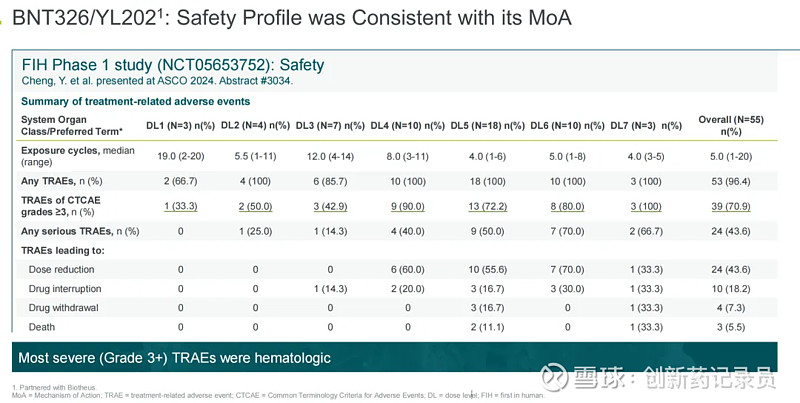
据LY202披露的临床数据,出现2例死亡的第五组剂量组中,入组了18位患者。这是目前该临床试验的7个分组中,收入患者最多的组别。
宜联生物联合创始人、首席运营官肖亮前两天接受媒体采访时表示,“高剂量下发生的安全性事件实际上是今年年初就产生的数据。”
第五组剂量分组(4.0mg/kg)里的18个人中,或许有部分是今年上半年加快入组的。
年中,宜联生物首席医学官在接受ASCO相关采访时,对外表示:研究人员最终选择低于 4.0 mg/kg 的剂量水平进行下一步剂量优化。
从安全事件发生,到年中对外公布数据,这个口径基本意味着,高剂量确实是影响安全性的重要因素。
那么,就此转战低剂量试验止损可以吗?
答案是不行。FDA需要进一步了解产品本身。有时候ADC与化药,不过一念之差。
众所周知,ADC产品的魔力来源于搭配组合:由特定的Linker将抗体和小分子毒素连结成为一个整体。产品经过静脉滴注进入人体后,由抗体带领找到肿瘤细胞相关的特定抗原,而后精准释放毒性,发挥抗癌作用。
YL202能够引发死亡,无疑是产品的作用流程出现了问题。
据披露数据,在入组的55名患者中,整体≥3级TRAE发生率较高为70.9%,主要是血液毒性反应。且在3.0mg/kg、4.5mg/kg以及5.5mg/kg剂量组中,≥3级TRAE发生率达到80%以上。
这类不良反应在化疗中常见,原因是化疗需将细胞毒性药物静脉输注到人体时,药物经血液大循环再抵达身体各处,它们攻击癌细胞的同时也攻击正常细胞。血液性毒性反应,便是攻击正常细胞产生的副作用。
YL202的主要不良反应如此,很可能是产品还没有抵达癌变部位,毒素就开始裂解了。
ADC产品中有一个名为DAR的参考值,它意味着抗体携带的小分子毒素量,毒素量越大,对肿瘤细胞的杀伤力就大。大名鼎鼎的DS-8201,DAR值是8,此前凭此数值在ADC中成为一代翘楚。
而YL202的DAR值也是8。从毒性上看,YL202不输DS-8201。
但如果这类产品进入人体后,没有到达肿瘤部位便随机裂解,这无异于对患者进行了一场高强度化疗。此次临床入组的基本都是经过标准治疗的后线患者,他们能否承受得了这种治疗力度,也需要考量在内。
临床失误的原因种类复杂,目前还需要等待宜联生物与FDA的多方验证。
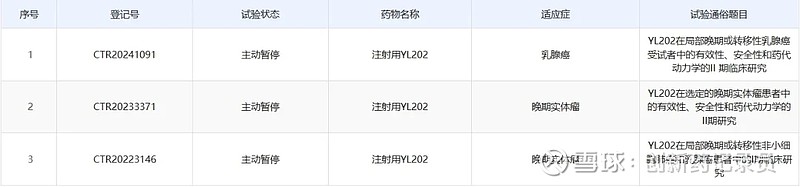
但在死亡病例的具体原因查明之前,这项临床只能先暂停。6月8日,该临床的美国临床入组暂停;11日,宜联生物对中国区临床也进行了主动暂停。
此事受关注度较高。市场记得宜联生物在Linker、抗体和毒素上的新见解,也期待了解此次临床死亡案例的真实原因。
$荣昌生物(SH688331)$ $百利天恒-U(SH688506)$ $复星医药(SH600196)$ #ADC创新药物# #创新药出海#