

药苑杂谈 YAOYUANZATAN
生物等效性,简称BE(bioequivalency),指在相似的试验条件下单次或多次给予相同剂量的试验药物后,受试制剂中药物的吸收速率和吸收程度与参比制剂的差异在可接受范围内。
按照研究方法评价效力的优先顺序为:药代动力学(PK)研究、药效动力学(PD)研究、临床研究和体外研究。
生物等效性研究是仿制药注册评价和上市药品一致性评价的重要研究手段,也是目前国际公认判断仿制药与原研药质量和疗效一致的金标准。
生物等效性研究法规汇总
小编通过检索NMPA、CDE官网,列出了目前现行有效的与BE相关的法规/指导原则,供研发人员参考。同时针对前段时间发布的BE摘要,以及21年发布的M9,小编也梳理了比较重要的信息,供大家参考。
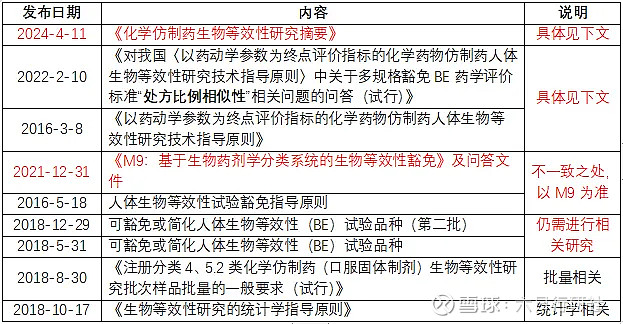
化学仿制药生物等效性研究摘要
发布日期:2024-4-11
发布内容:化学仿制药生物等效性研究摘要
发布要点:本摘要适用于以药动学参数为终点评价指标的生物等效性研究。申请豁免生物等效性研究的品种,可在生物等效性研究总结部分陈述豁免理由,并根据实际情况简化摘要撰写内容。采用药效学参数为生物等效性评价指标的研究,可参考本摘要撰写。仿制药质量和疗效一致性评价及其它涉及生物等效性研究内容的申请,相关内容可参考本摘要撰写。
《M9:基于生物药剂学分类系统的生物等效性豁免》及问答文件
发布日期:2021-12-31
发布内容:关于实施国际人用药品注册技术协调会指导原则《M9:基于生物药剂学分类系统的生物等效性豁免》及问答文件有关事项的通知
发布要点:1)NMPA不再单独发布可豁免或简化人体生物等效性试验品种目录,申请人评估认为符合M9指导原则要求的,相关的研究资料按照现行版《M4(CTD)》模块5.3.1.2项下统一提交,在药品注册申请中提出豁免人体生物等效性试验;2)《人体生物等效性试验豁免指导原则》与M9指导原则不一致的,以M9指导原则为准。
RA建议:申请人评估认为符合豁免要求的,建议准备相关研究资料提前与CDE进行沟通交流,如未进行沟通交流,经审评不能豁免的,注册申请不予批准。
CDE已公布的特药生物等效性研究技术指导原则
截至2024年6月20日,CDE共发布48条特药指南,小编梳理如下,供大家参考,如研发过程中遇到相关产品,可以下载原文遵照执行。
由于中国仿制药生物等效性试验评价体系起步较晚,特药指南的发布数量和频次跟FDA相比有不少差距,CDE目前已经发布的这些特药指南,是在积累的大量生物等效性研究数据的基础上,并主要参考FDA已公布的特药指南(少量参考欧盟),来逐渐完善的。
在这里提醒我们申请人,如国内仿制的品种,CDE暂未公布特药指南,但参比制剂来源于FDA或欧盟,则申请人有必要查看FDA和欧盟网站是否已公布相关特药指南,如有,应遵照执行。
预计未来,CDE会不断完善特药指南,并逐步赶上FDA和欧盟等先进药品监管机构的速度,为我国仿制药研发和一致性评价工作持续的推进打好基础。
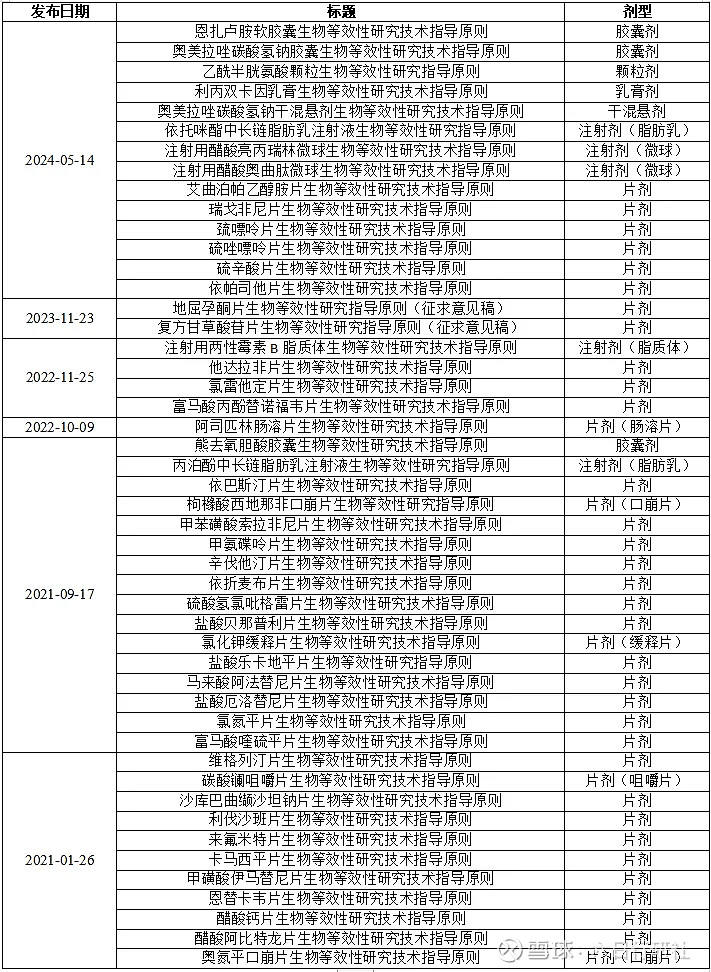
CDE 2016年发布的《人体生物等效性试验豁免指导原则》适用于仿制药质量和疗效一致性评价中口服固体常释制剂,以及按照新注册分类的化学仿制药,该指导原则又指出,基于BCS的生物豁免对治疗窗狭窄的药品和口腔吸收制剂不适用,所以在2020年的时候,CDE针对治疗范围狭窄的药品和口腔吸收制剂,发布了相关的指导原则。在2022年,CDE针对创新药也发布了相关指导原则。

CDE共性问题汇总
关于生物等效性问题,目前在CDE共性问题中可以检索到10条相关内容,具体信息小编总结如下,供大家参考学习。
1. 对于拟申请豁免生物等效性试验的品种,应根据《人体生物等效性试验豁免指导原则》,证明这些品种的BCS分类。对于BCS1类的品种,应进行溶解度、溶出度和渗透性研究,当辅料种类和用量与参比制剂不一致时,需提供数据证明辅料对吸收产生的影响;对于BCS3类药物,除需进行溶解度、溶出度研究外,还应处方种类一致,各组成用量相似。申请人应根据品种特点和自身资料按要求申报;
2. 对于BCS分类不明确药物,若想申请豁免BE,建议渗透性数据以人体数据为准,若原研说明书有相关数据,可以引用。人体数据不充分的情况下,体外渗透性数据可作为支持性数据。
3. 人体药代动力学研究是证明高渗透性最直观有效的方法,原研说明书的人体药代数据可作为药物渗透性的依据之一,当人体药代动力学数据不充分时,体外渗透性试验如Caco-2试验数据也可作为支持性证据。必要时,还应提供额外的文献资料进行佐证。
4. 基于ICH M9豁免生物等效性研究的化学仿制药,注册检验时应对多批自制制剂与参比制剂在多种溶出介质中进行体外溶出曲线对比复核。
5. 关于餐前和餐后BE,根据《以药动学参数为终点评价指标的化学药物仿制药人体生物等效性研究技术指导原则》,如果参比制剂说明书中明确说明该药物仅可空腹服用(饭前1小时或饭后2小时服用)时,则可不进行餐后生物等效性研究。对于仅能与食物同服的口服常释制剂,除了空腹服用可能有严重安全性方面风险的情况外,均建议进行空腹和餐后两种条件下的生物等效性研究。如有资料充分说明空腹服药可能有严重安全性风险,则仅需进行餐后生物等效性研究。
6. 关于BE备案规定:应提供三批中试以上规模自制样品的稳定性研究数据,试验样品包括但不仅限于中试放大批以及生物等效性试验批样品。应提供至少三个月加速与长期留样研究数据。在生物等效性试验期间继续进行以上样品进行加速与长期留样稳定性研究。如已拿到临床批件且在有效期内的,可不进行BE备案。
7. 关于BE批次,对于同一个试验,参比制剂和受试制剂应采用同一批次。
常见剂型BE梳理
1. 通常口服溶液、糖浆等溶液剂型(前提是辅料不会显著影响药物吸收或生物利用度时),可豁免BE,企业在立项调研时,需明确辅料是否会影响药物吸收,如有疑问,可以提前与CDE进行沟通交流;
2. 对于口服固体制剂(如片剂和胶囊剂),口服混悬剂、咀嚼片均需研究BE;
3. 对于不经肠道给药的溶液制剂:如静脉注射剂、滴耳剂、滴眼剂,如果自研制剂与参比制剂相比,原辅料完全一致,则可以豁免BE;
4. CDE在2018年发布过2次关于可豁免或简化人体生物等效性试验品种名单,如申请人仿制品种在所包含的名单里,仍需按照CDE 2016年发布的《人体生物等效性试验豁免指导原则》开展相关研究,建议与CDE进行沟通交流,评估符合要求的,在申报化学仿制药注册时,可以同时申请豁免或简化BE;
关于多规格口服固体制剂豁免BE的梳理
1. 如果各规格制剂在不同 pH 介质中体外溶出曲线相似,且各规格制剂的处方比例相似,通常可以基于最高规格的BE结果,采用体外药学评价的方法来豁免其他规格的BE;
2. 特殊情况,如最高规格有安全性方面风险,申请人可以基于以下三点要求:1)在治疗剂量范围内的线性药代动力学特征;2)最高规格与低规格之间的处方比例相似和溶出曲线相似;3)自研制剂与参比制剂溶出曲线相似,与CDE进行沟通交流,同意后,采用非最高规格的BE来豁免其他规格的BE;
3. 关于处方比例相似性,包括如下情况:1)不同规格之间所有活性组分和非活性组分比例完全相同;2)或者基本相同(即在处方等比放大或缩小的基础上,不同规格之间非活性成分的变化幅度在变更指导原则中的中等变更允许的±10%范围内),但这只适用于常释制剂;3)不相似或不完成相同,如申请豁免,需提供充分的理由与CDE进行沟通交流;
4. 对于高活性药物,即原料药在制剂中所占的比例小于5%,若不同规格使用相同的非活性组分,且各规格制剂重量差异不超过10%,则认为这种不同规格的高活性药物也属于处方比例相似。
往期回顾 Past Review