每年的10月1日是“国际戈谢病日”。作为一种危及生命的罕见遗传病,戈谢病并不为大众所知,全球确诊的戈谢病患者不足10000例,其中中国约有400例。“罕见但不孤独”,戈谢病理应得到大众的关注。研发客特约专栏作家梁贵柏应邀撰写此文,讲述戈谢病发现、诊断的历史以及特效药物思而赞的故事。
撰文 | 梁贵柏
1882年,法国一位名叫菲利普·戈谢(Philippe Gaucher)的医学院学生在毕业前的实习期间接触到了一个很特殊的病例。
这位32岁女性患者的脾脏肿大,开始被怀疑是得了癌症。她不幸去世之后,戈谢医生进行了病理解剖,发现她并非死于癌症。虽然直接死亡原因是败血症,但是该患者的内脏器官却呈现出多种特殊的表征,包括脾脏和肝脏中细胞增大,并伴有条纹状细胞质,等等。在随后撰写的毕业论文中,戈谢医生对这个患者的临床表现,以及病例解剖的结果做了详细的描述。

从那以后,与之相关的类似病例开始见诸于文献报道,从临床表现到家族的外观模式都有了更详细地描述。到了20世纪初,美国病理学家纳森·布里尔(Nathan Brill)医生在研究了家族病史后指出,这是一种遗传性的疾病,父母双方必须将致病基因传给孩子才会得病,并首次用“戈谢病(Gaucher’s Disease,简称GD)”这个名字的来描述这种罕见的遗传病。
布里尔医生还首次给活着的戈谢病患者做出了诊断。
追根寻源找病因
脾脏和肝脏的肿大是戈谢病最特异性的典型症状,有关戈谢病的研究也是从这里开始的。
1934年,一位法国化学家发现,导致戈谢病患者脾脏和肝脏肿大的原因是一种叫做葡萄糖脑苷脂(glucocerebroside)的脂肪物质在这两个器官中的积聚,这种脂肪物质的累积还会引起戈谢病的其它症状,例如血小板减少、贫血、疲劳、骨骼疼痛和病理性骨折等问题。
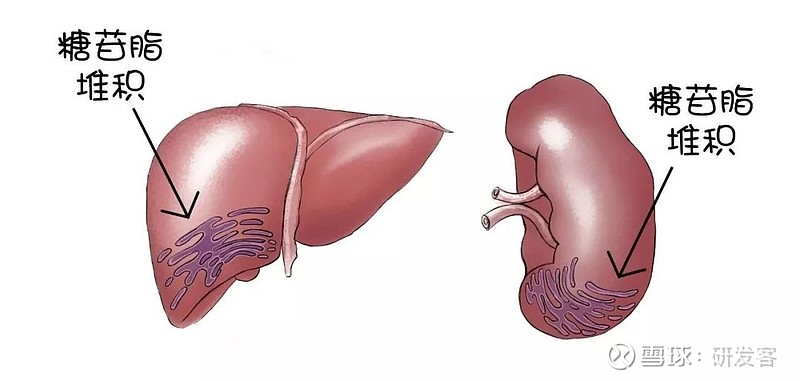
戈谢病患者肝脏和脾脏中的条纹状细胞质即糖苷脂堆积。
于是,专家们开始研究葡萄糖脑苷脂在人体内的代谢。1965年,美国生化学家布雷迪(Roscoe Brady)的研究团队发现,戈谢病患者体内葡萄糖脑苷脂的累积,不是因为葡萄糖脑苷脂的生物合成过量了,而是因为它的降解途径出现了问题。按图索骥,他们进一步发现,戈谢病患者缺少葡萄糖脑苷脂的主要降解酶—葡萄糖脑苷脂酶(glucocerebrosidase,也称为酸性β-葡萄糖苷酶)的活性只有正常人酶活性的10-20%,其中活性最高的地方是在细胞内的溶酶体中,因此戈谢病被称为是一种溶酶体贮积症。
很显然,戈谢病患者体内的葡萄糖脑苷脂酶发生了功能性缺损的变异,这就要在编码这种酶的DNA中找原因。果然,随后的分子生物学研究显示,戈谢病患者的第一条染色体中编码葡萄糖脑苷脂酶的基因发生了变异,导致了酶活性的下降。不同的变异还会导致不同类型的戈谢病I型、II型、或III型。
检测聚焦染色体
戈谢病的生化和遗传病理基础被揭秘之后,原先必须经历的复杂而又痛苦的骨髓穿刺检测诊断,被简单易行而且十分精准的血液检测和唾液基因检测取代了。
血液测试主要是分析葡萄糖脑苷脂酶的活性水平。根据酶的活性,医生可以判断疾病的严重程度。唾液基因检测则可以发现某些基因突变与类型I、II或III型戈谢病相关,从而诊断患者是哪种类型的戈谢病。布雷迪教授的团队还开发了一个产前测试,用以诊断胎儿的戈谢病。
现在通过基因测序我们还可以确定谁是这种疾病基因的携带者。携带者没有症状,却有可能将疾病传给他们的后代。
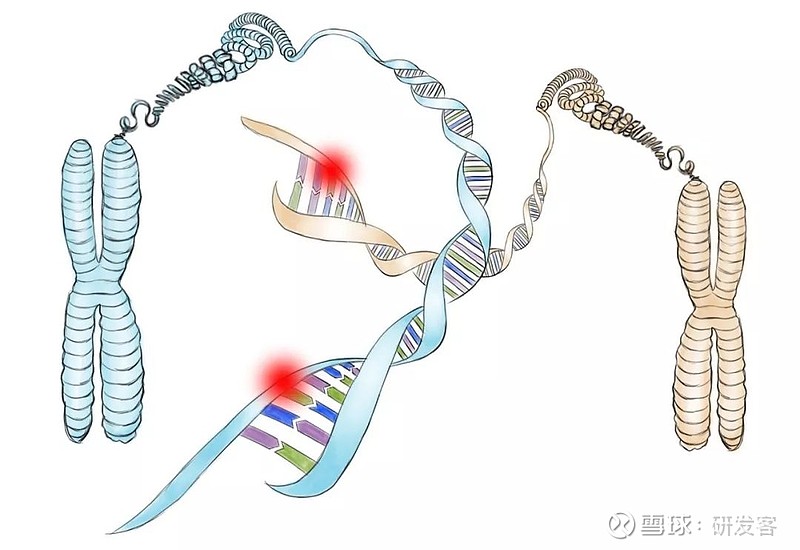
父母双方的缺陷DNA。随后的分子生物学研究显示,戈谢病患者的第一条染色体中,编码葡萄糖脑苷脂酶的基因(NDA)发生了变异。
目前已知的三种类型的戈谢病都是常染色体隐性。换句话说,人体内的两条染色体中只要有一条是正常的,这个人就不会出现戈谢病的症状,尽管他携带戈谢病的基因。只有当两条染色体都出现了戈谢病的变异,它们所表达的葡萄糖脑苷脂酶就一定会出现活性缺损,戈谢病才会发生。所以说,只有当父母双方都是携带者(不是患者,所以都有一条正常染色体)时,他们的后代才有可能患戈谢病,患病的几率为四分之一(25%)。每个胎儿从父亲和母亲各继承一条染色体,组成一个新的染色体对,只要其中有一条染色体不含有戈谢病的变异,疾病就不会发生。
戈谢病的致病基因在东欧阿什肯纳兹(Ashkenazi)血统的犹太人中携带者的比例要高得多,大约每十人就有一个携带者,所以患者比例也显著升高,大约每450个活产婴儿中就会有1个戈谢病患儿。
不容忽视罕见病
戈谢病是一种罕见的遗传病,全球病例不到10000例。根据2019年中国戈谢病治疗20周年媒体发布会的消息,中国目前确诊的戈谢病患者大约有400例,被列入中国官方2018公布的《第一批罕见病目录》。
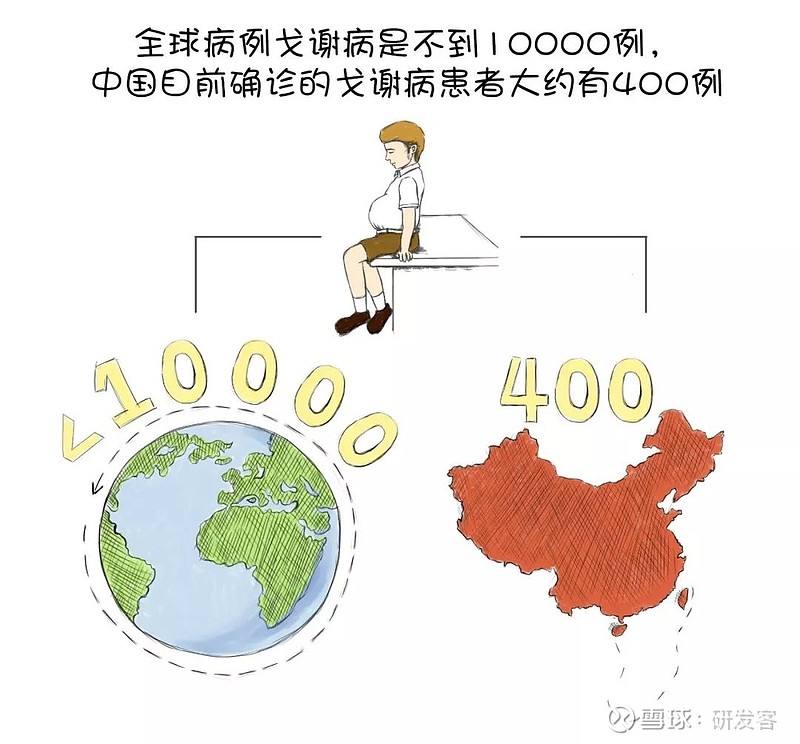
美国戈谢病基金会指出,在美国一般人口中,每100人中约有1人是I型戈谢病的携带者,发病率为4万分之一,属于罕见病(rare disease)。根据2002年美国国会通过的《罕见疾病法》,“影响美国20万人以下的任何疾病或状况”,或平均每1500人中出现1个病例可归类为“罕见病”,完全按照发病率划分。日本也是一样,罕见病的法律定义是影响日本不到5万名患者的疾病,即平均每2500人中有1个病例。
而欧盟罕见病的定义则包括了除了发病率以外的因素,如“危及生命、或慢性衰弱性疾病,其患病率如此之低,需要共同努力才能攻克的疾病”,一些统计上罕见但不危及生命、或治疗不足的疾病则被排除在罕见病的定义之外。
虽然不同的国家和地区对罕见病的定义有所不同,但是大家对戈谢病看法却是一致的,没有争议:这是一种危及生命、需要全社会的共同努力才能攻克的疾病。
约2/3的戈谢病患者是在儿童期发病,除了前面提到的肝脾肿大之外,其它主要症状为生长发育落后和贫血,往往还伴随着多器官功能性损伤,甚至会危及生命。
对症下药酶替代
然而,在戈谢病的生化和遗传病理基础被揭秘之前,医学界对戈谢病的治疗一直停留在“头疼医头、脚疼医脚”的初级状态,主要以减轻症状为目的。
戈谢病患者的脾脏肿大,医生建议的方法就可能是脾脏切除;患者的肝脏肿大,医生就会建议肝脏移植;骨骼和关节出了问题就做骨科的修复手术;血液或骨髓有了病变就实施血透或骨髓移植……其中,骨髓移植对少数I型戈谢病患者是有一定疗效的。
这些治疗手段对于减轻症状,也就是我们常说的“治标”,在短期内是有一定疗效的,但是它们都未能触及到戈谢病的“本”。
要想治本,就必须先弄清楚戈谢病的起因。在了解了戈谢病患者体内葡萄糖脑苷脂酶功能性缺损之后,医学界就把治疗方向转向了修复葡萄糖脑苷脂酶的功能,最直接的方法就是给戈谢病患者补充葡萄糖脑苷脂酶。
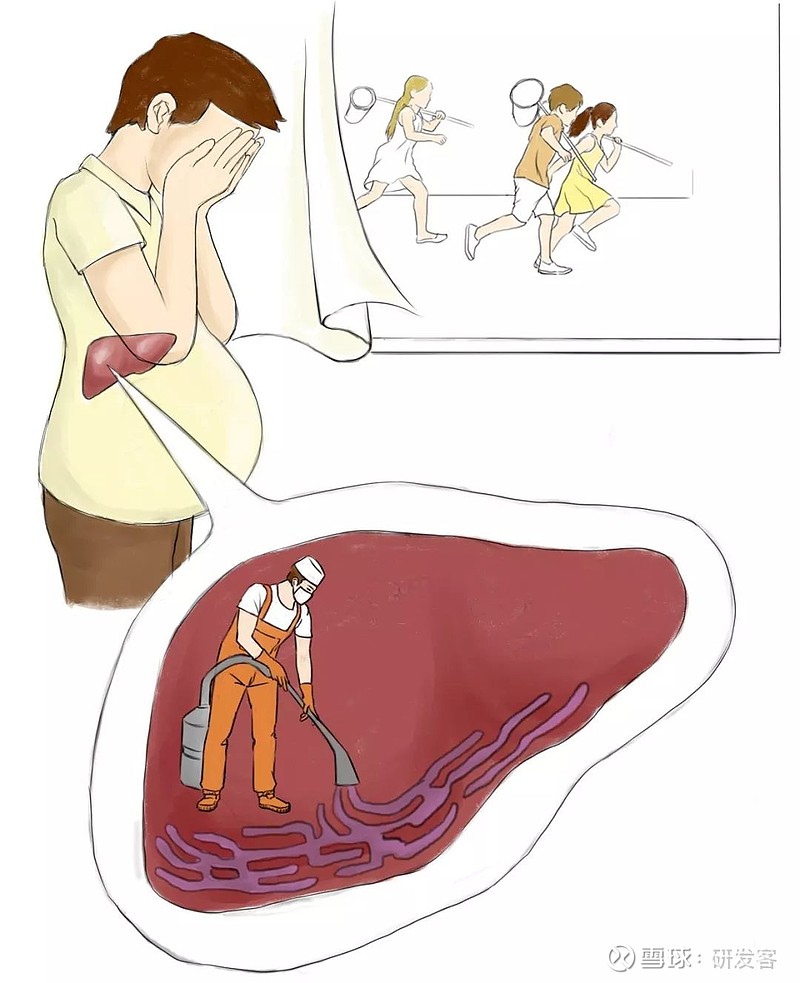
在了解了戈谢病发病机制后,医学界把治疗方向转向修复葡萄糖脑苷脂酶功能,通过葡萄糖脑苷脂酶清理肝脏里堆积的“垃圾”——糖苷脂。
但是在生物工程技术取得突破性进展之前的1960年代,分离和纯化足够量的葡萄糖脑苷脂酶用于治疗可不是一件容易的事。经过了多年努力,前文提到的布雷迪的研究团队终于积累了一定量的葡萄糖脑苷脂酶,并于1973年开始了临床试验。但是结果却不尽人意。
注射了葡萄糖脑苷脂酶之后,虽然有一部分戈谢病患者体内的葡萄糖脑苷脂开始下降,但是其它患者的响应率不高,观察不到疗效。
这是怎么回事呢?科学家们只好重新回到实验里继续研究。
修饰蛋白见成效
他们发现,葡萄糖脑苷脂酶必须进入到葡萄糖脑苷脂聚集的巨噬细胞内,才能对这些脂质体进行有效地降解,而葡萄糖脑苷脂酶本身并不能有效地进入巨噬细胞,它需要另一些分子的帮助,不幸的是,这些辅助分子在分离和纯化过程中都被除掉了。
能不能对葡萄糖脑苷脂酶进行改造,在保持它对脂质体降解活性的条件下,使得它也能被巨噬细胞识别,从而顺利进入巨噬细胞对脂质体进行有效的降解?
通过反复研究他们发现,这些巨噬细胞对甘露糖(Mannose)“情有独钟”,不但可以识别带有甘露糖残基的蛋白质(酶是蛋白质的一种),而且还会将这些蛋白质转运进入细胞内。于是他们用化学方法除掉了天然葡萄糖脑苷脂酶上一些寡糖残基,使它的外表呈现甘露糖的残基,希望巨噬细胞能够识别,并将这个酶转运进入细胞内。
他们成功了,靶向巨噬细胞的葡萄糖脑苷脂酶(macrophage-targeted glucocerebrosidase)诞生了。
在随后进行的酶替代治疗(Enzyme Replacement Therapy,简称ERT)的临床试验中,大部分患者的主要症状都有了明显的改善。在接受治疗的几个月之内,他们的肝脏和脾脏都开始缩小,身高和体重开始正常增长,血液和骨骼等多项指标也开始正常化。
1991年美国FDA批准了第一代酶替代药物上市Ceredase,戈谢病终于有了有效的药物治疗。
来之不易祖孙缘
这个来之不易的药物是由生物技术公司健赞(Genzyme,现为赛诺菲子公司)从婴儿出生后的胎衣(Placenta)中提取的。由于含量极低,治疗一个成年戈谢病患者所需的药物,必须每年从数百吨的人胎衣中提取才能获得足够的量。
1981年,美国哈佛大学、麻省理工学院和国立健康研究院的几位大牌教授强强联手,成立了健赞公司,专攻生物制品和药物,第一个重头项目就是酶替代药物Ceredase。
当时,人胎衣中提取的主要生物制品是免疫球蛋白。每年,世界上大约三分之一的婴儿出生后的胎衣,经过处理之后都被送往法国里昂市,集中进入了位于的赛诺菲巴斯德(当时为巴斯德梅里埃,Pasteur Mérieux,Rhône-Poulenc的子公司)的组织库。巴斯德的科学家在完成了免疫球蛋白的提取之后,再把这些胎衣组织的残留物转运到位于波士顿郊外的剑桥市的健赞公司,进行天然糖苷酶的提取,用于制造酶替代药物Ceredase。。
从人胎衣残留物中提取糖苷酶可不是一件容易的事, 考虑到获得人胎衣原材料的国际规模前所未有,每年涉及到世界各地数千吨的人胎衣及时处理、贮藏、运输等许多环节,糖苷酶的成功提取和Ceredase的稳定生产真的是一个不小的奇迹。
在健赞公司的创始人里,有一位是麻省理工学院的分子生物学教授Harvey Lodish,为健赞的成功做出了很重要的贡献。但是他怎么也没有想到,多年以后,自己的小孙子竟然得了戈谢病,自己家族的基因里就带着戈谢病的变异。值得庆幸的是,因为他自己和同行们的一起努力,戈谢病已经有药了,小孙子得到了及时治疗,已经长成高挑的帅小伙了。
健赞创始人Harvey Lodish与患戈谢病孙子的合影。
生物工程思而赞
1970年代后期,生物工程技术的突破性进展为蛋白质修饰和表达提供了很大的方便,研究人员可以在实验室里快捷地从天然蛋白质分子上除掉或者换掉他们不想要的基团,也可以加上另一些他们设计好的基团,然后用基因重组技术来表达和生产蛋白质。
所以健赞公司在提取天然糖苷酶和生产Ceredase的同时,也开始探索用全新的基因重组技术来表达和生产葡萄糖脑苷脂酶,从而摆脱对于人胎衣的依赖,获得更安全、质量更稳定的酶替代药物。
他们对实验室里培养的中国仓鼠卵巢细胞(Chinese hamster ovary cell)做了基因工程的改造,植入糖苷酶的基因,并成功地表达出了新型的葡萄糖脑苷脂酶。
与化学制品不同,生物制品组成是随着生产工艺的变化而变化的。虽然这个重组的葡萄糖脑苷脂酶与从人胎衣中提取的天然葡萄糖脑苷脂酶出自同一个基因编码,但是由于表达细胞系和生物环境的不同而不完全相同,它们的免疫原性也会有所不同,所以必须经过严格的安评与生物等效性评价。
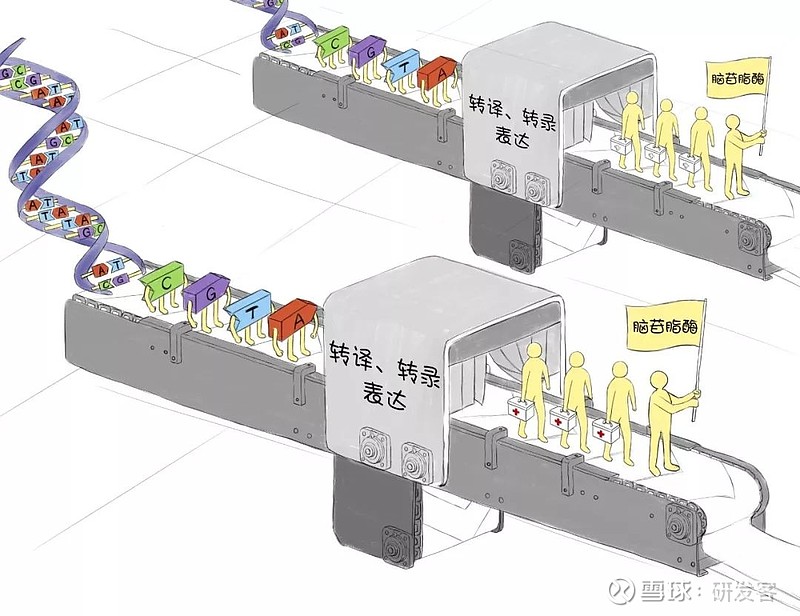
在生物工程生产线上,DNA编码从一端输入,另一端转化成脑苷脂酶。生物工程技术极大的提高了第二代戈谢病酶替代药物的产量和稳定性,也大大降低了成本。
直接头对头的临床试验结果显示,这个重组的葡萄糖脑苷脂酶与Ceredase相比,在体内的性质以及药效上没有显著区别,在抗体反应(~20%)方面则略低于Ceredase(~40%)。
1995年,通过生物工程技术生产的第二代治疗戈谢病的酶替代药物注射用伊米苷酶(lmiglucerase,商品名Cerezyme,思而赞)上市了。
同舟共济戈谢病
2009年,思而赞在中国被批准上市,是目前国内唯一获批的戈谢病特异性酶替代药物治疗手段。

虽然现代生物工程技术的应用极大地提高了产量和稳定性,同时也降低了部分生产成本,但是思而赞仍然价格不菲。面对一个非常小的患者群体(全球不足10000例),思而赞商业回报的考量是很有挑战性的。
研发新药需要天文数字的长期投入,而商业回报又是保证大量资源投入的前提。对于人数比例极少的罕见病来说,仅仅依靠患者个人及其家庭的支付能力是不可能达到供需平衡的,必须有全社会的帮助和投入。
1999年,赛诺菲开始与世界健康基金会合作,为中国134名戈谢病患者提供无偿药品援助。2009年思而赞在中国上市后,又获得了来自中华慈善总会的帮助,由他们与赛诺菲联合实施援助项目。这是中国第一个针对罕见病的慈善援助项目,到目前为止已经积累了10年以上的戈谢病诊疗的真实世界数据。
据2019年中国戈谢病治疗20周年媒体发布会的消息,到目前为止该项目共发放援助药品思而赞57025支,价值逾14亿人民币 ,帮助了134名戈谢病患者接受治疗。通过治疗,完全实现生活自理的患者比例达到61.5%,不需要辅助用具比例接近80%,处于劳动和学习状态的高达85%。他们重新获得了健康,过上了正常人的生活,甚至能为家庭和社会贡献自己的力量。
2019年9月于上海
主要参考文献
Patrick B. Deegan and Timothy M. Cox, “Imiglucerase in the treatment of Gaucher disease: a history and perspective” Drug Design, Development and Therapy, 2012, 6, 81–106.
Wikipedia, Gaucher’s disease, last edited on 22 August 2019.
National Gaucher Foundation office website: 网页链接

梁贵柏
梁贵柏博士曾在默沙东新药研究院工作多年,潜心钻研药物化学,颇有建树。几年前回国加入药明康德,从事业务开发、项目管理和驻美运营。梁博士是《新药研发的故事》一书的作者。他以长期的积累、独特的视角和生动的文字,通过《老梁说药》栏目讲述新药研发“背后的故事”,令人耳目一新,脑洞大开。
梁贵柏博士目前是偕怡制药联合创始人兼首席科学家,欢迎业界读者通过邮箱gbliang55@hotmail.com与梁博士联系。
责编 | 张咏晴 姚嘉
绘图 | 杨予晴
总第864期
访问研发客网站可浏览更多文章
