(备注:文章太长,超过雪球允许的2万字,一分二/OYang)
(备注:这是疫情至今,对于新冠新冠重症最全面最有说服力的回顾性论文,刚好与舒泰神的发明专利和 #补体药物# C5a在其中的关键作用,就翻译了过来。)
上篇:网页链接
宿主内在的机制
鉴于单核细胞缺乏ACE2的表达或表达较弱,血液单核细胞被新冠病毒感染是令人惊讶的,这表明有其他病毒进入机制的参与20,58。体外健康捐赠者单核细胞的感染被抗尖锐湿疣抗体或患者康复期血浆明显增强,这表明单核细胞通过抗体依赖性吞噬作用摄取了被消化的病毒颗粒58。事实上,病人血浆的抗体消耗和阻断单核细胞Fc受体FcγRIIIa(也称为CD16)的抗体在很大程度上废除了单核细胞感染。虽然这一发现与另一项研究的结果相矛盾122,但在严重的新冠肺炎患者中看到的afucosylated抗病毒抗体增加的观察结果支持了这一点(参考文献123,124)。IgG抗体的Fc结构域包含一个保守的N-连接聚糖,在这个聚糖上缺乏核心岩藻糖基化的IgG对FcγRIIIa的亲和力更高。因此,岩藻糖基化可能启动增强的抗体依赖性吞噬,导致单核细胞感染和炎症体激活。无论病毒进入途径如何,单核细胞是促炎症细胞因子的主要贡献者,并在新冠病毒感染后发生热化现象,这表明它们可能是新冠肺炎中严重炎症的重要驱动因素。
除了新冠病毒直接感染介导的炎症体激活外,炎症体在新冠肺炎中被间接激活也是可信的(图1)。通过各种机制,包括宿主翻译抑制,新冠病毒抑制早期I型干扰素反应,使病毒复制不受控制125,126,127。溶解受感染的肺细胞可以分泌报警素来激活炎症巨噬细胞并促进它们被招募到肺部128。通过过量表达新冠病毒 E蛋白裂解的Vero E6细胞的上清液可以诱导受体巨噬细胞分泌IL-1β119,可能是通过NLRP3,但也可能是其他炎症体传感器,如AIM2和NLRC4(参考文献58)。有趣的是,与NLRC4和NLRP3表达增加有关的eQTL与严重的新冠肺炎相关,表明可能有多个炎症体传感器参与该疾病58。
激活新冠肺炎中炎症体的宿主内在机制也可能涉及先天或适应性淋巴细胞。细胞毒性T淋巴细胞和自然杀伤细胞可以通过传递颗粒酶A,裂解气蛋白B(GSDMB)或颗粒酶B,裂解GSDME,导致孔的形成77,129,从而诱导感染细胞发生热化。鉴于GSDMB在气道上皮细胞中的高表达,细胞毒性淋巴细胞介导的对感染细胞的杀伤可能有助于肺部感染期间细胞因子和DAMP的释放129,130。此外,病原体诱导的裂解补体产物C5a,在严重的新冠肺炎患者的BALF中含量很高,可以促进MERS-CoV感染小鼠的IL-1β释放,也可以通过CD4+T细胞的ROS依赖性机制引发NLRP3的激活109,131,132。
另一个也许更有病理意义的ROS依赖性机制涉及氧化通常由肺泡上皮分泌的富含磷脂的表面活性剂,它异常地产生氧化磷脂,是小鼠感染模型中ARDS和树突状细胞和巨噬细胞中非经典炎症体(人类caspase 4/5或小鼠caspase 11)的有力诱因33,133,134。这些现象共同表明对DAMPs、补体产物和局部肺泡液的复杂反应,产生有利于炎症体激活的肺部微环境,导致炎症细胞因子和报警素的释放,肺上皮细胞和组织驻留的巨噬细胞过度激活,通过热化作用造成组织损伤,以及随后外周免疫细胞的促炎症性招募。
因此,通过新冠病毒感染的直接、细胞自主激活和/或DAMPs的间接激活,NLRP3等炎症体传感器可以作为新冠肺炎致病机制的关键多因素驱动因素。支持这一观点的令人信服的证据也来自蝙蝠,它们与对人类具有高度致病性的冠状病毒无症状地共存。蝙蝠的外周血单核细胞表达NLRP3的低态异构体,因此在MERS-CoV感染后不能释放IL-1β;然而,这种功能通过表达人类NLRP3得以恢复,这表明NLRP3的抑制可能是减少冠状病毒致病性的有效策略106。
GSDMD、NETosis和凝血功能障碍
与SARS-CoV和H1N1流感病毒感染一样,新冠病毒可以引起炎症导向的凝血病,这可能是新冠肺炎中静脉和动脉血栓的高发率和高死亡率的原因(图2)。凝血病的标志是血浆凝血标志物的水平增加,如D-二聚体、因子VIII和Von Willebrand因子,以及凝血酶原时间的异常9,135,136,137。虽然已知促炎症细胞因子可促进各种促凝血因子,但它们可能不足以导致一些严重的新冠肺炎患者出现戏剧性的全身性血凝块形成。单核细胞或中性粒细胞中的炎症体激活GSDMD也可以通过多种机制促进凝血。炎症体的激活和热化作用可以以一种不依赖细胞因子的方式引发大量的凝血病。在内毒素血症的小鼠模型中,通过caspase 1或caspase 11激活炎症体信号,导致全身纤维蛋白沉积,让人联想到弥散性血管内凝血,这一现象取决于GSDMD,而不是IL-1β或IL-18(参考文献138,139)。GSDMD依赖性裂解高温巨噬细胞,释放出富含膜结合组织因子的细胞外小泡,这是一种跨膜糖蛋白,在血液中循环,通过激活凝血酶促进凝血功能138,140。来自新冠肺炎患者的血浆胞外囊泡显示组织因子活性增加,这也与疾病的严重程度相关140。炎症体诱导的凝血病也可能涉及磷脂酰丝氨酸介导的组织因子的激活。磷脂酰丝氨酸通常被限制在内膜小叶中;然而,Ca2+通过GSDMD孔进入,激活磷脂扰动酶跨膜蛋白16F(TMEM16F),使磷脂酰丝氨酸外化以增强组织因子活性141。
图2:炎症体驱动的新冠肺炎的机制。
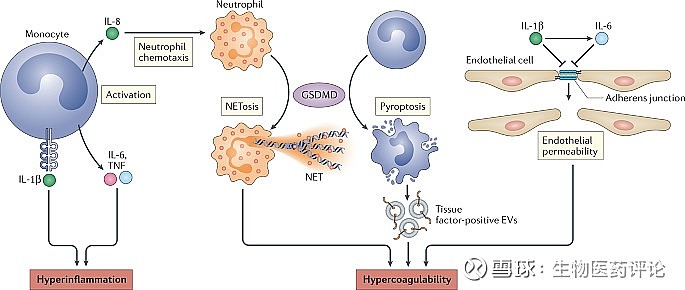
炎症体信号释放的IL-1β激活了单核细胞,单核细胞分泌IL-6、肿瘤坏死因子(TNF)和IL-8。这些细胞因子通过各种机制引起炎症,包括招募中性粒细胞到肺部。中性粒细胞中Gasdermin D(GSDMD)的激活导致中性粒细胞胞外陷阱(NETs)的形成,它可以招募血小板并促进高凝血能力。IL-1β和IL-6可以下调内皮细胞的粘附连接,这增加了它们的通透性,可能有助于肺部血管的凝结。由高温单核细胞释放的组织因子阳性细胞外囊泡(EVs)也可以直接激活凝血级联,促进新冠肺炎的凝血。
在中性粒细胞中,GSDMD是形成中性粒细胞胞外陷阱(NETs)的必要条件,NETs是由装饰有抗菌蛋白的DNA组成的挤出的纤维状组合142,143,144。在病毒感染期间,NETs会促进凝血功能失调,鉴于新冠肺炎中大量中性粒细胞浸润肺部毛细血管,这一点特别值得关注(参考文献145,146)。新冠肺炎患者血液中的循环中性粒细胞显示NETs的基础激活增加,而且NETs在肺部也高度丰富147,148,149。NETs招募血小板并将其纳入微血栓中。因此,GSDMD对凝血和NETs的激活可能在新冠肺炎的炎症期间变得失调,从而导致在严重疾病中看到的弥散性血管内凝血。炎性体激活同时驱动新冠肺炎的严重炎症和凝血的可能性表明,抑制它可能是有益的。
临床干预措施
炎症体激活和IL-1β活性在许多慢性炎症中失调,并已成功地成为药物的目标,其中许多药物目前正被重新用于治疗新冠肺炎(参考文献28,150)。Anakinra是人IL-1RA的重组和略微修改的版本,在小型队列研究中显示了早期的前景,但临床试验的结果参差不齐28,150,151,152,153,154,155,156。一项试验由于没有明显减少通气需求和死亡率而提前停止155。值得注意的是,这项研究只招募了轻度至中度疾病的患者,其C反应蛋白水平截止值略微升高,不需要入住重症监护室,可能排除了对抗IL-1治疗反应最强烈的人155。其他免疫调节剂也有类似的考虑,如地塞米松,其死亡率的降低仅限于接受呼吸系统支持的人14,157。另一项试验使用了可溶性尿激酶浆细胞激活剂受体的水平,这是严重新冠肺炎的一个强有力的早期预测指标,并发现每天服用安纳金拉能显著减少进展到严重呼吸衰竭(危险比0.3)和30天死亡率(危险比0.49)156,158。然而,由于这是一项单臂研究,需要进行安慰剂对照的随机试验。抗IL-1干预的成功率参差不齐,这可能表明IL-1不依赖的焦化作用促成了疾病的进展,这是单一细胞因子调控所不能达到的。
多个II期临床试验正在对轻度或重度新冠肺炎患者进行NLRP3直接抑制试验(诺华公司,NCT04382053;Olatec Therapeutics公司,NCT04540120)(表1)。在动物模型中,NLRP3抑制剂可减少A型流感病毒感染的细胞因子释放和肺部炎症159,NLRP3抑制剂MCC950可减少体外感染新冠病毒的初级人类单核细胞的Caspase 1激活和IL-1β的分泌52。NLRP3的另一种抑制剂,即磺酰脲类糖尿病药物甘伯瑞,减少了新冠病毒感染的人类单核细胞的IL-6分泌65。这些数据表明,与抑制IL-1相比,抑制NLRP3可能是对抗新冠肺炎的一个优越策略。间接抑制NLRP3的方法也在试验中,即使用微管解聚药物秋水仙碱,秋水仙碱的其他作用是一种耐受性良好的炎症体形成抑制剂160,161。在中度至重度患者中进行的一项小型随机对照试验表明,它能够减少氧气需求和住院时间;在这项研究中,由于总死亡率低,无法确定它对死亡的影响162。另一项研究表明,用秋水仙碱治疗的新冠肺炎患者的住院率或死亡合并率明显下降,尽管住院率和死亡率本身都没有明显下降,该研究还没有经过同行评审163。糖尿病药物二甲双胍通过调节mTOR间接抑制NLRP3,其使用与新冠肺炎和2型糖尿病患者的死亡率降低明显相关。最近,二甲双胍也被证明可以抑制NLRP3的激活和新冠病毒感染小鼠的肺部炎症164,165。
表1 正在进行和计划进行的抑制炎症体途径的临床试验新冠肺炎
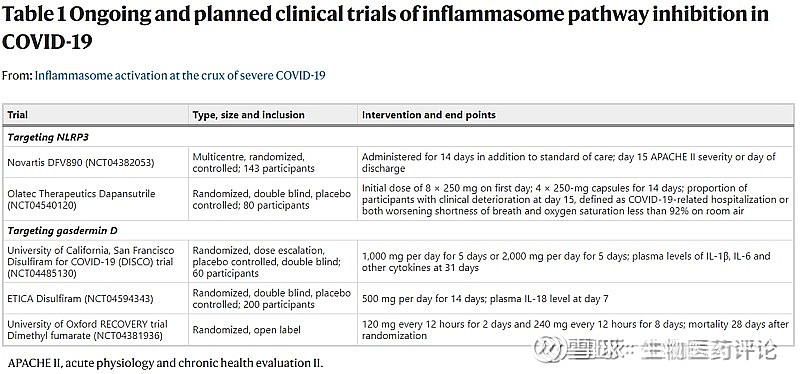
GSDMD是另一个有吸引力的目标,因为孔的形成发生在所有炎症体传感器的下游,但在IL-1β和DAMP释放的上游166。最近有两种已知的小分子药物被描述为可抑制GSDMD。双硫仑(DSF)是美国食品和药物管理局(FDA)批准的治疗酒精依赖的药物,使用了70年,通过共价修饰Cys191来抑制GSDMD孔的形成,并保护小鼠免受LPS引起的败血症和炎症细胞因子的分泌167。富马酸二甲酯(DMF)是FDA批准的治疗多发性硬化症的药物,以及富马酸盐,与DMF有关的内源性克雷布斯循环中间体,最近被证明可以抑制GSDMD168。有趣的是,几个多发性硬化症患者接受DMF治疗的病例系列显示,他们的新冠病毒感染是自限的,而他们没有接受任何特定的治疗169,一项使用大型卫生保健数据库的观察性研究显示,与对照组相比,使用DSF治疗酒精中毒的群体感染新冠病毒的风险明显降低,而且没有死亡,但这项研究还没有得到同行的全面审查170。目前,DSF正在两个对照的II期临床试验中进行研究,这些试验正在测试轻度至中度疾病(NCT04485130)以及住院病人(NCT04594343)的疗效(表1)。同样,DMF正在一项随机开放标签试验中测试其对新冠肺炎患者死亡的预防效果(NCT04381936)(表1)。DSF和DMF也可能通过其抗病毒活性赋予治疗新冠肺炎的有益效果。DSF可弱化抑制新冠病毒 MPro多聚蛋白蛋白酶171。新冠肺炎患者显示出核因子红细胞2相关因子2(NRF2)抗氧化基因的表达受到抑制。NRF2激动剂4-乌头酸辛酯(一种细胞可渗透的乌头酸衍生物)和DMF都激活了NRF2依赖的抗病毒程序,以抑制新冠病毒在肺上皮细胞系中的复制172,173。
当人们考虑使用抗炎药物时,一个重要的问题是何时是干预的最佳时机。早期,炎症通过招募免疫细胞到感染部位并激活它们的扩张和保护功能,促进保护性适应性免疫的发展,过早干预可能会干扰有效免疫的发展。后来,前馈机制接管了,干预措施可能无法控制炎症的爆炸性放大及其危险后果。因此,"甜蜜点 "可能是肺部窘迫的迹象刚刚开始变得明显的时候(例如,血氧饱和度轻微下降、轻度呼吸困难和胸部X射线的早期肺炎证据)。需要注意的是,大多数症状轻微的病人不会发展成严重的疾病,而检测严重疾病终点的改善需要大量的研究参与者。总的来说,治疗时机和适当的病人分层将是确定抑制炎症体的干预措施和一般的抗炎药物是否有益的重要参数。
结论性意见
最近的数据支持炎症体参与严重的新冠肺炎,通过直接感染介导的激活或间接DAMP介导的激活。炎症体信号和IL-1β的释放对多种病原体的保护作用已被证实,特别是在感染的急性阶段174,175,176。然而,其长期的晚期激活可能是免疫病理学的基础,包括过度的细胞因子释放、伴随免疫细胞浸润的肺内皮损伤和全身高凝状态。在早期新冠病毒感染的情况下,炎症体信号是否具有保护作用,有待于使用体内疾病模型进一步研究,而用炎症体抑制剂治疗的时机可能是关键。
还值得提醒的是,NLRP3是一个杂乱地激活的感染和压力传感器,它的慢性激活一般不会导致在一些新冠肺炎患者身上看到的严重炎症(参考文献30)。病程的变化可能与炎症体激活刺激的程度或传播、炎症的部位或发炎的细胞类型有关。事实上,大鼠的经典实验表明,IL-1β在肺部的特异性过表达足以再现ARDS的许多表型177。炎症体诱导的病理变化也可以用限制慢性炎症体信号下游的反馈放大的负性调节机制来解释。同时激活炎症体和抑制炎症体的负调控机制可能会产生严重的不可控炎症。值得注意的是,对抑制和终止炎症体激活的反馈机制仍然知之甚少。
未来的研究需要解决I型干扰素信号和炎症体激活的相互抑制问题,以及这两个先天免疫信号系统之间的相互作用如何导致新冠肺炎的严重性(图3)。这两种主要的先天性免疫反应常常通过不完全了解的机制相互拮抗,在许多情况下是什么决定了哪种反应占主导地位也不完全清楚178、179、180。新冠病毒感染抑制早期干扰素信号,同时激活炎症体信号52,58,91。I型干扰素通路基因的功能缺失突变和I型干扰素的自身抗体与严重的新冠肺炎有关(参考文献181,182),而年龄进一步抑制I型干扰素反应183。在新冠病毒感染中,病毒内在和宿主内在对干扰素反应的抑制应损害抗病毒免疫力,但也可能通过消除炎症体活动的这一负向调节器而导致细胞因子释放不受限制。无节制的炎症体激活可以驱动严重的疾病并发症。例如,缺乏炎症体活性的I型干扰素缺陷小鼠,尽管有类似的病毒滴度184,185,但对流感病毒感染相关的并发症的敏感性较低。早期给药的重组I型干扰素不仅可能恢复先天的抗病毒免疫反应以更好地控制新冠病毒,而且可能同时抑制随后的高炎症。然而,临床试验还没有证明I型干扰素有明显的益处。虽然有效和特异的抗病毒药物应该比免疫调节药物做得更好,但只有一种抗病毒药物被批准186。尽管如此,炎症体信号的过度激活和热化作用越来越可能是新冠肺炎发病机制的驱动因素,需要进行控制性的临床研究来评估药物再利用以抑制炎症。
图3:新冠肺炎中早期I型干扰素和IL-1β的相互抑制。
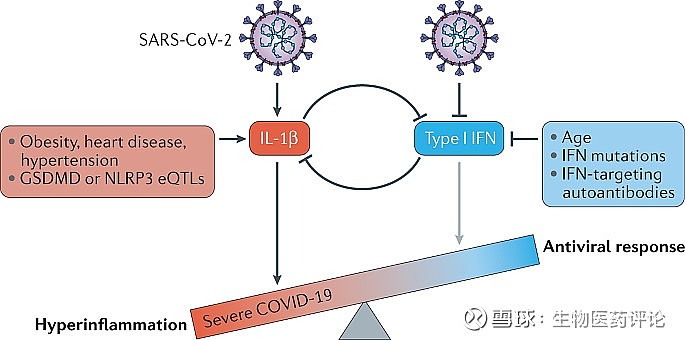
严重急性呼吸道综合征冠状病毒(新冠病毒)感染的早期阶段促进IL-1β的分泌,同时抑制I型干扰素。各种严重疾病的易感性与基础IL-1β产生的增加有关(如肥胖、心脏病、高血压和与增加的gasdermin D(GSDMD)或NLRP3水平有关的表达定量性状位点(eQTLs))或I型干扰素产生的减少有关(如年龄、干扰素途径突变和自身抗体),这可能抵消这两个信号系统之间的平衡,将固有免疫反应推向高炎症状态,同时抑制抗病毒反应。IFN,干扰素。
------------------------------------------------------------------------------------------
备注:文章太长,引用无法放得下。
详见原文:网页链接