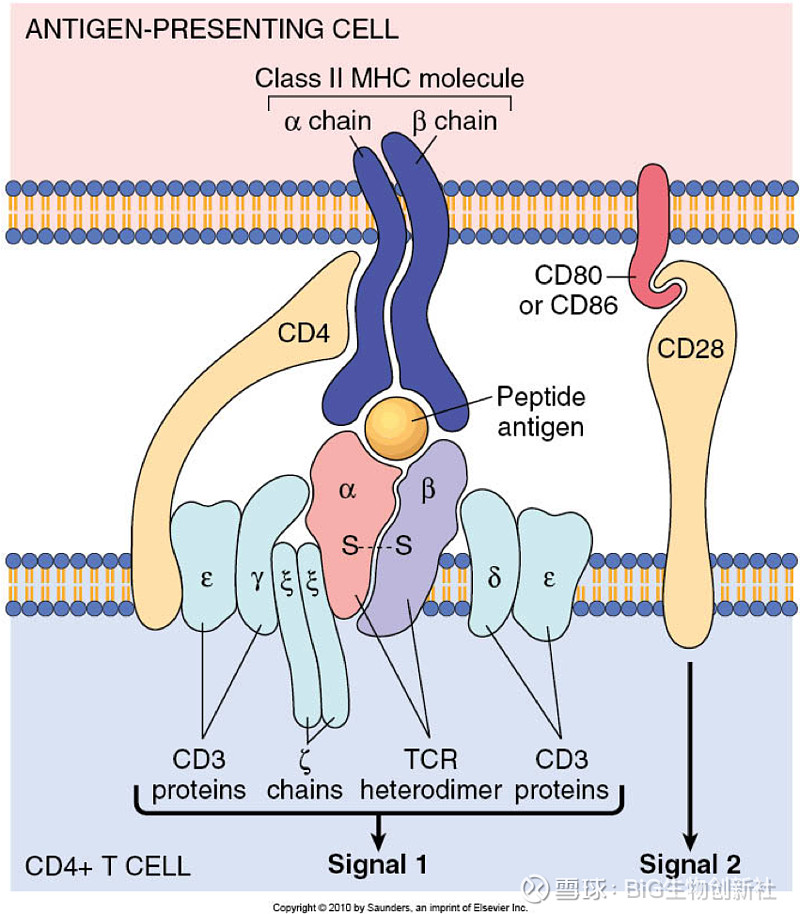
T细胞受体、共刺激信号和细胞因子信号协调地支配触发T细胞激活和功能编程的特定信号网络。了解免疫代谢信号网络的调节器和效应器可能会发现调节人类疾病中代谢程序和T细胞反应的治疗靶点。
在这篇文章中,我们总结了丝氨酸/苏氨酸激酶介导的关键信号网络的上游调节器和信号效应器,包括调节代谢的PI3K–AGC激酶、mTOR和LKB1–AMPK途径,特别是在T细胞中的调节作用。我们重点讨论了参与免疫代谢调节的细胞信号通路,主要涉及参与代谢调节的相关分子、上游和下游靶点及其细胞类型特异性效应,分为PI3K、AGC、mTOR的机制靶点和LKB1–AMPK信号。
当T细胞受体(TCR)在共刺激和细胞因子信号存在的情况下识别同源抗原时,原始T细胞中的信号网络被激活,以促进克隆扩增、效应细胞分化和免疫功能。在抗原清除后,大多数效应细胞通过程序性细胞死亡而死亡,但一些细胞作为记忆细胞则会长期存在,并为快速回忆进行下一次免疫斗争做好准备。此外,T细胞的功能状态有着相同和不同的转录程序,以及蛋白质表达、活性和相互作用的差异。在现阶段的研究中,这些差异才刚刚开始被理解,人们也越来越认识到细胞代谢在决定T细胞发育和功能方面的重要性。

PI3K-AGC signaling
磷脂是影响下游免疫代谢途径的关键第二信使,因此在转换方面受到高度调节。磷脂转换的一个中心介质是PI3K,它将磷脂酰肌醇-(4,5)-二磷酸(PIP2)转化为磷脂酰肌醇-(3,4,5)-三磷酸(PIP3)。PIP3的产生引起质膜募集和含有PH(pleckstrin homology)结构域的蛋白质功能调节。
因此,PI3K活性可以通过将大量含有PH结构域的效应蛋白招募到邻近区域来产生亚细胞信号中枢。PI3K活性诱导参与调节细胞功能的多种信号通路,包括Akt(蛋白激酶B)、磷脂酰肌醇依赖性蛋白激酶1(PDK1)以及mTOR复合物1。
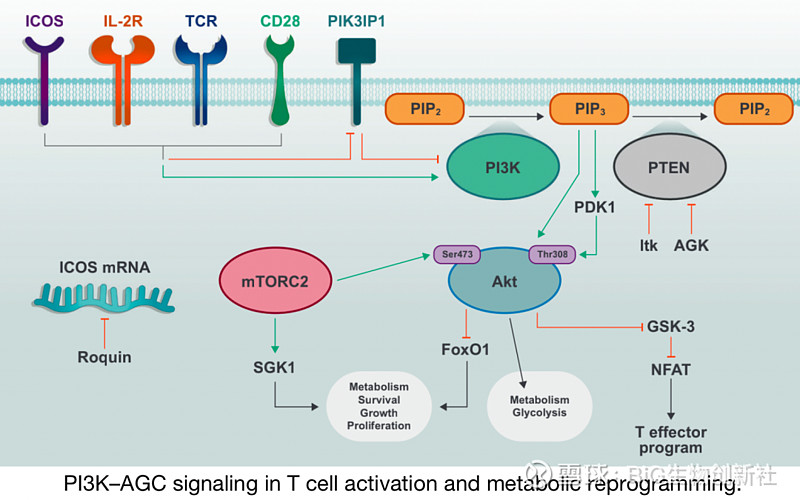
PI3K–T细胞活化和代谢重编程中的AGC信号
TCR、CD28和IL-2R的激活诱导PI3K的磷酸化和激活,以及PI3K抑制分子的失活。PIP2通过PI3K的活性转化为PIP3,PIP3促进包括PDK1和Akt在内的下游信号分子的质膜募集和激活。mTORC2进一步激活Akt并促进代谢增加和T细胞效应器功能。
1.1
PI3K
《Protein kinase C: perfectly balanced》
I类PI3K是一种异二聚体蛋白质,由一个催化的p110亚单位(p110α、p110β、p110δ或p110γ)和五个调节亚单位亚型(p50α, p55α, p55γ, p85α, or p85β)中的一个组成。在没有刺激的情况下,催化亚单位的激酶结构域被调节亚单位抑制。PI3K底物的浓度与细胞激活状态密切相关,在T细胞中,细胞激活状态由TCR结合、CD28家族介导的共刺激和细胞因子信号(如IL-2R下游)决定。在上游激活后,PI3K的Src-homology-2(SH2)结构域可结合上游受体或衔接蛋白上的特异性磷酸化YXXM基序,导致调节亚单位的抑制性连接释放和催化亚单位移位至富含底物的质膜。
PI3K信号由磷酸酶活性负调控。具体而言,PIP3分别通过磷酸酶和张力蛋白同系物(PTEN)以及含SH2结构域的肌醇5′-磷酸酶(SHIP)转化为PIP2或PI-(3,4)-P2。SHIP在促进Th1细胞反应中起着至关重要的作用,而PTEN缺乏会导致T细胞过度活化,尤其是在次优刺激下。PTEN在强TCR刺激或CD28共刺激下被抑制,部分原因是Tec家族激酶IL-2诱导的T细胞激酶(Itk),并且在CD8 T细胞激活期间也受到AGK介导的磷酸化的负调节。
1.2
AGC激酶
在最显著的含有PH结构域的蛋白质中有AGC激酶的成员,包括PDK1、Akt、核糖体S6激酶(RSK,也称为p90)和血清/糖皮质激素调节激酶1(SGK1)。PDK1是一种丝氨酸/苏氨酸激酶,在激活其他AGC激酶(包括Akt和SGK1)中起重要作用。TCR和CD28激活促进PDK1向质膜的募集并诱导其磷酸化, Thr513处PDK1的蛋白激酶C-θ(PKC-θ)依赖性磷酸化,而不是激酶结构域残基(Ser241)的自动磷酸化,这对于驱动T细胞活化至关重要。
在活化的CD8 T细胞中,PDK1参与代谢重编程,维持IL-2刺激下游的葡萄糖摄取和糖酵解,该过程需要mTOR–HIF-1α(缺氧诱导因子-1α)轴,但不需要PI3K或Akt活性。PDK1活性的适当调节对于加强适当的T细胞活化以及控制炎症的Treg细胞功能至关重要。在Ser473处的mTORC2磷酸化后,Akt达到最大激活,这允许Akt结合到被称为PDK1的“PIF口袋”的底物dock,这促进了Thr308处Akt的磷酸化。mTORC2–Akt信号则可以协调T细胞激活、分化和运输。
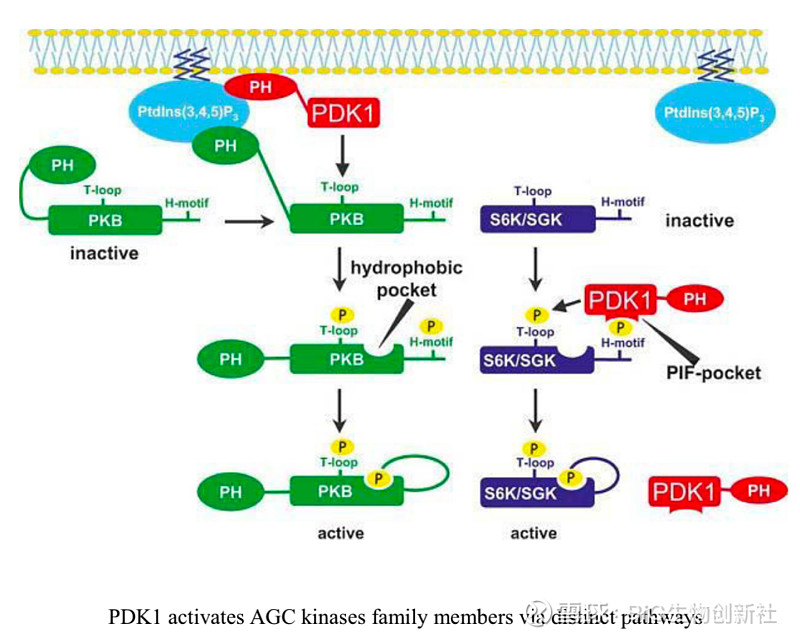
《The Clinical Implications of the Survival Pathway in Prostate Cancer》
Akt在T细胞中的一个重要作用是通过磷酸化调节叉头盒O(FoxO)转录因子的活性,这导致它们被排除在细胞核之外并终止靶基因转录。FoxO蛋白在静止的细胞群中更具转录活性,它们通过KLF2促进稳态细胞因子受体IL7R的表达以及细胞运输分子(如CD62L、CCR7和S1PR1)的表达。最近的证据表明FoxO1活性的适当下调对T细胞内环境稳定至关重要。从机制上讲,活化T细胞中的FoxO1下调对于协调细胞生长和增殖非常重要,因为它允许持续的mTORC1信号和合成代谢,因此,Akt–FoxO1轴动态调节T细胞反应。
另一种参与Akt信号传导的丝氨酸/苏氨酸激酶是糖原合成酶激酶3(GSK-3),它在静息细胞中具有组成性活性,但通过Akt的磷酸化而失活。活性GSK-3在T和B细胞中通过限制活化T细胞的核因子(NFAT)活性,促进其存活并限制活化。AGC激酶(如RSK)及其底物在T细胞代谢规划中的确切作用一直备受关注,也同样是目前研究的重点方向。

mTOR signaling
mTOR是一种丝氨酸/苏氨酸蛋白激酶,存在于两种信号复合物mTORC1和mTORC2中。mTORC1由mTOR激酶定义,mTORC1的适配蛋白调节相关蛋白和SEC13蛋白8(MLST8)的哺乳动物致死蛋白,以及脯氨酸Akt底物-1(PRAS1)及其靶DEP结构域相互作用蛋白(DEPTOR)的抑制亚单位。mTORC1信号对胸腺中的T细胞发育、外周的内环境稳定和分化为效应CD4 Th1、Th2和Th17细胞以及细胞毒性CD8 T细胞至关重要。相比之下,Th1和Th2细胞分化更需要mTORC2活性,同时后者也能调节Tfh和Treg细胞的迁移。
mTOR信号通过调节体内Treg细胞的激活、谱系稳定性和抑制功能,在对抗传统T细胞反应中发挥额外作用。最后,mTOR信号可拮抗淋巴组织中长期记忆CD8 T细胞的分化,却可促进其在非淋巴组织中的发育。因此,mTOR信号是T细胞反应的中央调节器。
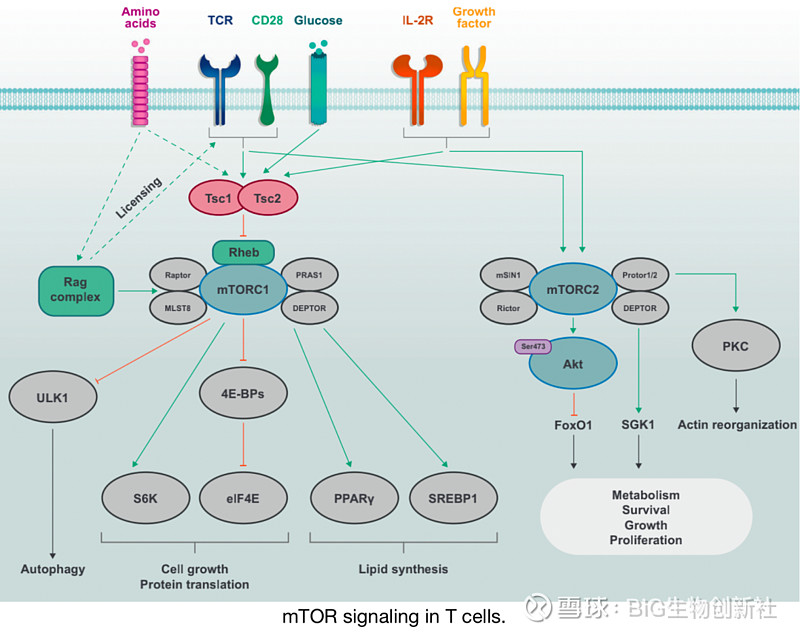
T细胞中的mTOR信号
离散的mTOR复合物mTORC1(由mTOR、Raptor、PRAS1、DEPTOR和MLST8组成)和mTORC2(由mTOR、Rictor、Protor1/2、mSIN1和DEPTOR组成)由免疫受体(TCR、CD28和IL-2R)和生长因子激活。
mTORC1的激活也对氨基酸等营养素敏感,氨基酸通过Rag复合物促进mTORC1激活,Rag复合物也起到允许TCR和CD28共刺激信号诱导mTORC1激活的“许可”作用。Tsc复合物的活性受到免疫和生长因子信号的抑制,通过抑制小G蛋白Rheb的激活,后者促进mTORC1的激活。mTORC1通过S6K和eIF4E诱导细胞生长和蛋白质翻译,并通过PPARγ和SREBP1诱导脂质合成。在营养充足的条件下,mTORC1通过ULK1抑制自噬。mTORC2主要在细胞生长、增殖和存活的环境或亚特异性代谢重编程中起着关键作用。

LKB1-AMPK signaling
LKB1–AMPK信号通路在调节细胞代谢、增殖和生存以应对营养和能量需求的改变方面起着核心作用。LKB1–AMPK信号可促进产生ATP的分解代谢途径,并使T细胞在应对能量应激时具有代谢可塑性。通过调节代谢重编程,LKB1和AMPK有助于T细胞的分化和功能。在以下部分,我们将介绍LKB1和AMPK活性是如何调节的,其对代谢的影响以及在T细胞介导的免疫中的作用。
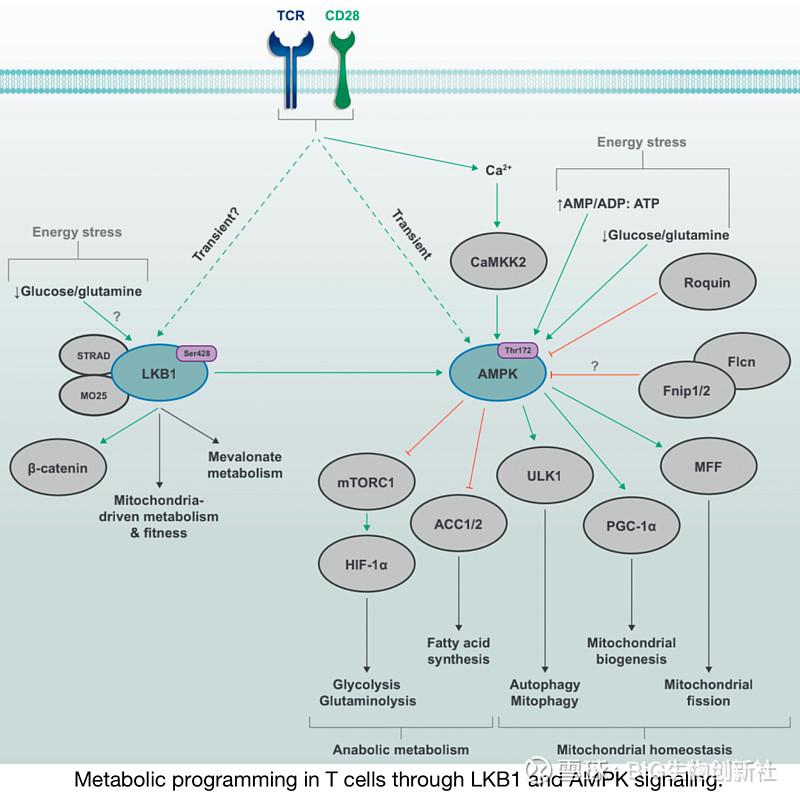
通过LKB1和AMPK信号在T细胞中进行代谢编程
能量应激途径激酶LKB1和AMPK由TCR和CD28共刺激信号激活,AMPK活性部分由Ca2+CAMMK2途径介导。在缺乏葡萄糖或谷氨酰胺或AMP/ADP/ATP比值失衡,也可促进LKB1–AMPK信号传导。上游营养感应蛋白,如Fnip–Flcn复合物和Roquin,可以抑制AMPK功能。LKB1的激活与线粒体代谢和适应度的变化以及特定环境下甲羟戊酸代谢的增加有关。通过调节多个下游靶点的活性,AMPK信号可阻止糖酵解、谷氨酰胺解和脂肪酸合成的代谢程序,同时促进分解代谢过程,如有丝分裂吞噬和自噬。AMPK还通过促进线粒体凋亡来驱动线粒体生物发生和线粒体动力学,从而支持线粒体活性。
3.1
LKB1和AMPK的调节

《The LKB1-AMPK pathway: metabolism and growth control in tumour suppression》
LKB1是一种丝氨酸/苏氨酸激酶,具有肿瘤抑制功能,参与调节细胞代谢和增殖。LKB1有许多下游靶点,包括定义最明确的靶点AMPK和AMPK相关激酶,如BRSK、NUAK和MARK。LKB1的上游调节由细胞定位和翻译后修饰介导。LKB1与STE20相关适配器(STRAD)和MO25形成复合物,分别促进LKB1的细胞质定位和激酶活性。此外,Akt可以通过促进LKB1核滞留来抑制LKB1。
AMPK是一种保守的丝氨酸/苏氨酸激酶,由α、β和γ亚基组成。AMP和ADP与CBS3重复序列的结合限制了磷酸酶对AMPK催化α亚基上Thr172的访问,该亚基是促进AMPK激酶活性的关键磷酸化位点。最终,代谢产物的复杂调节允许AMPK活性在不同能量应激条件下的“分级”反应。至少三种上游激酶介导AMPK Thr172磷酸化:LKB1、钙钙调素激酶激酶2(CaMKK2)和TGF-β激活激酶1(TAK1),其中LKB1和CaMKK2的作用最为清楚。
在T细胞中,TCR和Ca2信号以及CD28共刺激协同作用,通过CaMKK2快速、瞬时激活AMPK。AMPK活性也通过蛋白质-蛋白质相互作用进行调节。Fnip1和Fnip2与AMPK形成复合物,Fnip1的功能缺失突变提高了B细胞的AMPK活性,从而确立Fnip1是AMPK的负调节因子。Fnip结合蛋白Flcn也与AMPK相互作用,可能作为AMPK信号的负调节因子,有证据表明Fnip1和Flcn受AMPK的相互调节。
3.2
LKBI-AMPK介导的分解代谢
AMPK活性与低营养丰度诱导一致,通常与分解代谢程序有关,以生成ATP。AMPK通过ULK1的磷酸化激活自噬,ULK1可促进自噬介导的线粒体内稳态,促进T细胞存活。此外,AMPK独立于mTOR调节,在葡萄糖受限时,AMPK还通过Ser15处p53的磷酸化介导细胞周期阻滞。因此,LKB1–AMPK信号通过限制能量应激期间的合成代谢和细胞生长促进细胞存活。
此外,通过磷酸戊糖途径产生的代谢物核酮-5-磷酸对LKB1–AMPK途径的破坏促进了脂肪生成。相反,LKB1也可以促进甲羟戊酸代谢,以AMPK非依赖性方式支持Treg细胞内稳态。因此,LKB1–AMPK信号主要作用于抑制脂质合成途径,但AMPK依赖性LKB1信号可能在选择性情况下促进新生脂肪生成。
对肿瘤细胞的研究表明,LKB1或AMPK信号的破坏促进有氧糖酵解,部分是通过HIF-1α、因此增加糖酵解酶的转录。HIF-1α的LKB1–AMPK依赖性调节也可能部分依赖于mTORC1的抑制。LKB1–AMPK信号可能通过HIF-1α或ACC1介导的糖酵解和线粒体氧化代谢变化间接协调Th17和Treg细胞系的分化。此外,最近的研究表明,LKB1促进稳定的Foxp3表达,以及Th2样Treg细胞的发育,独立于AMPK和mTORC1–HIF-1α信号,但依赖于β-连环蛋白信号。LKB1信号是TCR介导的Treg细胞激活后线粒体功能和线粒体依赖性代谢程序所必需的,包括FAO或嘌呤和嘧啶代谢。这些发现强调LKB1和AMPK协调代谢重编程以调节T细胞分化和Treg细胞功能。

免疫代谢信号网络
上述主要信号通路在功能上并不排斥,相反,它们相互交织,相互作用,共同达到适当的代谢调节,以满足细胞功能的特定环境需求。因此,许多这些调节输入汇聚在共同的节点上,包括单个分子和整个细胞过程。
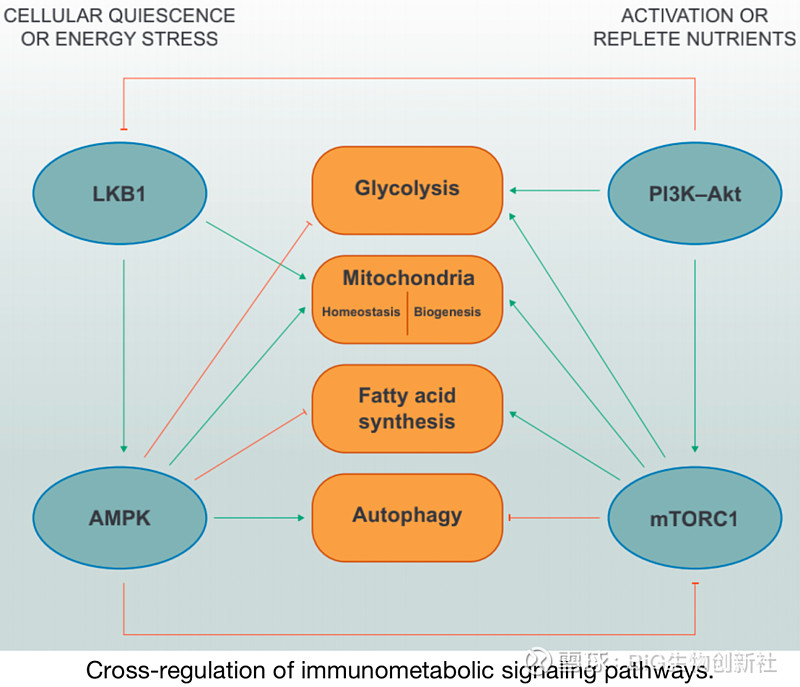
免疫代谢通路的交叉调节
在营养缺乏的条件下,LKB1–AMPK信号抑制合成代谢相关程序,如糖酵解和脂肪酸合成,同时促进线粒体内稳态和自噬。AMPK通过其专一性转接器蛋白Raptor的磷酸化直接抑制mTORC1。在激活和/或营养充足的条件下,PI3K–Akt和mTORC1信号促进糖酵解、线粒体生物发生和脂肪酸合成,同时抑制自噬。据报道,Akt可以磷酸化LKB1以抑制其功能定位。
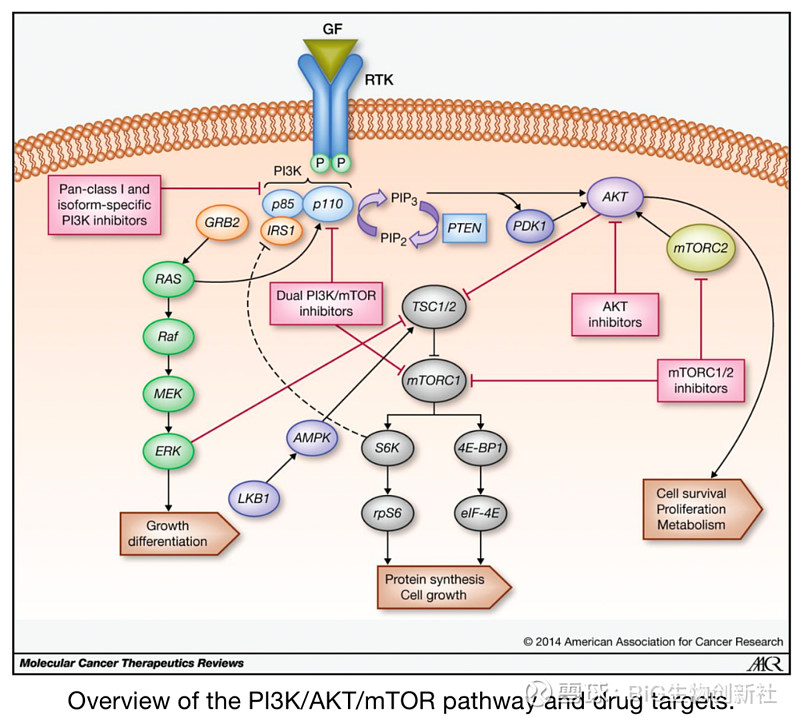
一般来说,PI3K–AGC激酶和mTOR活性的升高,以及LKB1–AMPK活性的降低,与细胞生长、增殖和效应器功能相关,这与代谢程序的变化相关。上游复合体的信号整合最终与下游代谢程序的调节有关,决定免疫细胞代谢程序的信号通路也受到代谢物和营养物质的交互影响。这种“双向代谢信号”为激酶依赖性信号和基因转录提供了另一层调控,以控制不同环境下的免疫细胞功能。
免疫信号网络在免疫代谢中的作用是一个令人兴奋的研究领域,对治疗和人类健康具有广泛的意义。在这篇综述中,我们强调了PI3K–AGC、mTOR和LKB1–AMPK信号在T细胞功能和命运中的关键作用。该领域在理解这些信号分子是如何调节的以及它们是如何重新编程T细胞代谢方面取得了很大进展,确定相对知名的上游免疫信号(如TCR、细胞因子受体)如何在代谢中传递不同的功能程序,可能为改善过继性T细胞治疗(如抗肿瘤CAR-T细胞治疗)提供新的治疗靶点。
参考文献:
Saravia, J., Raynor, J.L., Chapman, N.M. et al. Signaling networks in immunometabolism. Cell Res 30, 328–342 (2020). 网页链接
* 推文用于传递知识,如有版权等疑问,请于本文刊发30日内联系BiG生物创新社。