
最近这几年有个行业在逐渐地崛起,我们叫它医药合同外包,它代表的是从临床前到新药生产这一条产业链,目前产业链中有10来个上市公司,我们耳熟能祥的有药明康德、泰格医药、凯莱英等等,分别代表了前中后三个阶段。这里我们不仅好奇,作为研发能力薄弱的国家,外国为什么愿意把研发外包到中国来,而且趋势也越来越往中国集中了。
这里不得不提一下礼来曾经的阿兹海默症特效药研发失败的事件,花费了数十年的时间,投入了几十亿的美元最后化为乌有,对公司不仅是信心的打击,也是股价的打击。美国FDA的严苛是出了名的,在美国进行新药生产,不仅需要高端人才、顶尖技术和设备、也还需要足够多的样本和试错机会,这每一项都成本高昂。反观中国,人口教育红利以及产业升级的红利正在释放,最有竞争力的莫过于国内的样本数量和成本优势,强如十大也不得不勒紧钱包考虑把新药研发外包。
下面来从整个研发流程来看看,为什么新药研发又难时间又久。![]()
新药研发流程
一、新药研究筛选阶段
研究阶段包括四个重要环节,即靶标的确定,模型的建立,先导化合物的发现,先导化合物的优化。
1, 靶标的确定
在基础研究中,科学家们努力寻找特定疾病中发生作用的细胞和基因,以及针对特定生物参数和功能的化学或生物物质,希望能够发现其具有类似药物的作用。保守估计目前所有的药物治疗大概只覆盖了约700个药物靶标,在未来最少还有近10倍左右的药物靶标未被发现。药物的靶标包括酶、受体、离子通道等。
目前,较为新兴的确认靶标的技术主要有两个。
一是利用基因重组技术建立转基因动物模型或进行基因敲除以验证与特定代谢途径相关或表型的靶标。这种技术的缺陷在于,不能完全消除由敲除所带来的其他效应(例如因代偿机制的启动而导致的表型的改变等)。
二是利用反义寡核苷酸技术通过抑制特定的信使 RNA 对蛋白质的翻译来确认新的靶标。例如嵌入小核核糖核酸(snRNA)控制基因的表达,对确证靶标有重要作用。说的通俗一些,药物靶标的发现是基础科学的范畴,有必然性,但也许偶然性会更大一些。
2,模型的建立
靶标选定以后,要建立生物学模型,以筛选和评价化合物的活性。通常要制订出筛选标准,如果化合物符合这些标准,则研究项目继续进行;若未能满足标准,则应尽早结束研究。一般试验模型标准大致上有:化合物体外实验的活性强度;动物模型是否能反映人体相应的疾病状态;药物的剂量(浓度)——效应关系,等等。可定量重复的体外模型是评价化合物活性的前提。近几年来,为了规避药物开发的后期风险,一般同时进行药物的药代动力模型评价(ADME评价)、药物稳定性试验等。

3,先导化合物的发现
新药研制的第三步是先导化合物的发现。所谓先导化合物(leading compound),也称新化学实体(new chemical entity,NCE),是指通过各种途径和方法得到的具有某种生物活性或药理活性的化合物。因为目前的知识还不足以渊博到以足够的受体机制指导药物设计以使药物的合成不必使用预先已知的模型,所以,先导化合物的发现,一方面有赖于以上两步所确定的受体和模型,另一方面也成为了整个药物研发的关键步骤。一般来说,先导化合物主要有如下几个来源:对天然活性物质的挖掘、现有药物不良作用的改进以及药物合成心中间体的筛选等。目前,主要有两个获得新的先导化合物的途径。一是广泛筛选,这种毫无依据的方法在实际操作上其实是比较有效的。另外,先导化合物的合理设计近年来也越来越成为这一领域的热点。所谓合理设计,是指根据已知的受体(或受体未知但有一系列配体的构效关系数据)进行有针对性的先导化合物设计,这种方法有别于一般普遍筛选的显著特点在于目的性强,有利于各种构效理论的进一步发展,因此前途十分广阔。
先导化合物的广泛筛选,必须提一提高通量筛选。请看下图:
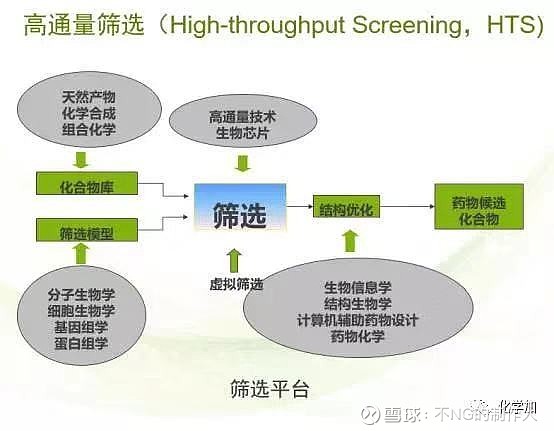
高通量筛选指以分子水平和细胞水平的实验方法为基础,以微板形式作为实验工具载体,以自动化操作系统执行试验过程,以灵敏快速的检测仪器采集实验结果数据,以计算机分析处理实验数据,在同一时间检测数以千万的样品,并以得到的相应数据库支持运转的技术体系,它具有微量、快速、灵敏和准确等特点。简言之就是可以通过一次实验获得大量的信息,并从中找到有价值的信息。
新分子一边合成出来,一边做各种生物活性,药代动力学,毒理这类的研究,找到我们需要的分子。
药动学参数:

毒性研究包括两类:急性毒性和长期毒性
急性毒性是指机体(动物)一次性或短时间内(24小时)多次接触外源性化合物后短期内所产生的毒性效应。
长期毒性是指机体长期连续或反复接触外源性化合物后所产生的毒性效应。长期毒性一般包括一周或两周,更长时间包括4周,8周甚至数年。早期安全性评价只做一周或两周的动物试验。
长毒试验通过剂量爬坡获得动物对该药物的最大耐受剂量(maximum tolerated dose,MTD)。毒性试验主要观察给药后动物的体重、摄食摄水量、症状、死亡、血液生化、肝活性、尿液以及动物出现死亡时进行组织病理切片。药物化学家根据早期安全性评价的结果,确定是否进一步优化先导化合物。
4,先导化合物的优化
在该阶段,生物学家和药物化学家一起努力使先导化合物更安全更有效。这是基于各步骤知识复杂和反复的过程。通常一个或多个先导化合物进行安全评价,合成和筛选。这就是所谓的类似物。类似物测试,其结果是与化合物结构变化相关的生物活性和药理数据。这些用于建立结构 - 活性关系(SAR)。新类似物将反馈到系统的下一个优化步骤中。最终得到优化的先导化合物,进入到临床前研究。进入临床前研究的化合物称为临床前候选化合物。
总结:到这一步才算是临床前开发准备工作的完成阶段,其实算是一个大量筛选目标化学物分子的工作,由上万到几千,由几千到几百,剩下几百个入选化学物分子再进入临床前和临床研究。整个的研发关键在临床研究上。
二, 新药开发阶段
新药开发阶段如下:临床前试验,研发中新药申请,临床研究,新药申请。
临床前试验:由制药公司进行的实验室和动物研究,以观察化合物针对目标疾病的生物活性,同时对化合物进行安全性评估。这些试验大概需要3.5年的时间。
研发中新药申请(Investigational New Application,IND):在临床前试验完成后,公司要向FDA提请一份IND,之后才能开始进行药物的人体试验。如果30天内FDA 没有发出不予批准的申明,此IND 即为有效。提出的IND需包括以下内容:先期的试验结果,后续研究的方式、地点以及研究对象;化合物的化学结构;在体内的作用机制;动物研究中发现的任何毒副作用以及化合物的生产工艺。另外,IND必须得到制度审核部门(the Institutional Review Board)的审核和批准。同时,后续的临床研究需至少每年向FDA提交一份进展报告并得到准许。然后就是临床一期,临床二期,临床三期以及上市后的安全性监督研究。

1, 临床前试验
临床前研究的主要内容:合成、生产和控制(Chemical、Manufacture & Control),制剂(Pharmaceutics),药理学(Pharmacology),药效动力学(Pharmacodynamics),药代动力学(Pharmacokinetics),毒理学(Toxicology),急性毒性试验(acute toxicity testing),重复给药毒性试验(repeat dose toxicity testing),长期毒性试验(long term toxicity testing),致癌性试验 (carcinogenicity toxicity testing),生殖毒性和致畸性(reproductive toxicity),基因毒性试验/致诱变性(genotoxicity/mutagenicity),毒代动力学(toxicokinetics)等。这里面的每一项内容都需要大量的时间和金钱的投入。
2, 研发中新药申请
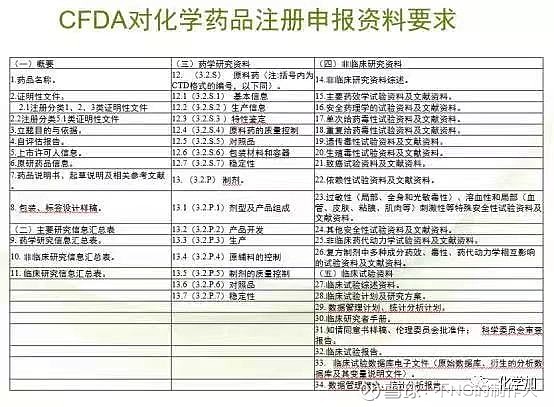
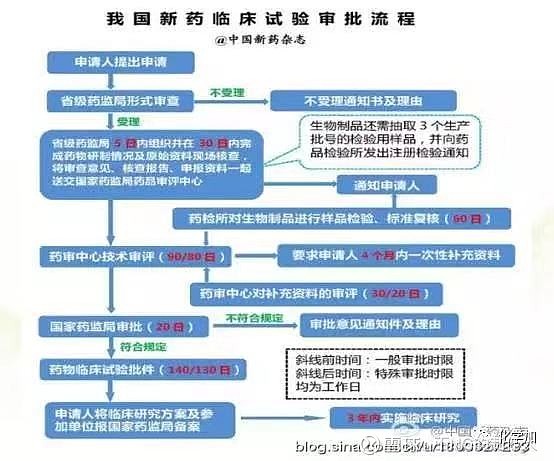
3, 临床研究
临床研究一般包括四个阶段,即,首次试用于人类(健康受试者)的一期临床研究,试用于少数患者的二期临床研究,有上千患者参加的三期扩大临床研究,以及上市后进行实用验证的四期临床研究前三期为新药上市前必经阶段,第四期为药品上市后的监督性研究。
新药临床试验的分期及基本要求:
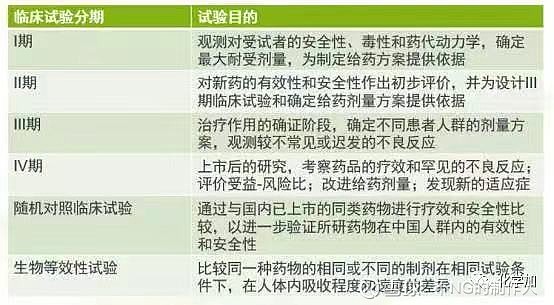
l期临床试验,一般为20~30例,有时会到100例。
II期临床试验,多中心临床试验,一般试验组不少于100例。
III期临床试验,更大规模的多中心临床试验,要求试验组不少于300例。如为双盲试验设计,则至少为300例;如为单盲试验设计(试验组和对照组并非1:1的比例),则只需满足试验组不少于300例。
IV期临床试验,不少于2000例。随机对照临床试验,多中心临床试验,一般不少于100对。如为多个适应症,则每个主要适应症的病例数不得少于60对。生物等效性试验(BE试验),一般为18~24例。根据药物的特性,可适当调整样本量的大小(变异越大的药物所需的病例数越多,目前国外最多有做到100多例的BE试验)。
4, 新药申请
通过临床试验,公司将分析所有的试验据。如果数据能够成功证明药物的安全性和有效性,公司将向FDA提出新药申请。新药申请必须包括公司所掌握的一切相关科学信息。典型的新药申请有10万页甚至更多。根据法律FDA审核一份NDA的时限应该为6个月。但是几乎所有案例中的新药申请从首次提交到最终获得FDA批准的过程都超过了这个时限;在1992年对于新分子实体的新药申请平均审核时间为29.9个月。

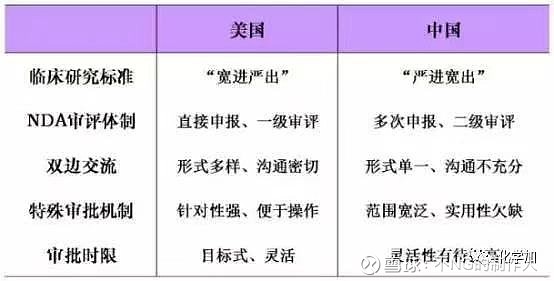
关于新药研发有一点不得不说一下,就是中国和美国在相关流程上有不同之处:
5、批准上市
一旦FDA批准新药申请后,该药物即可正式上市销售,供医生和病人选择。但是还必须定期向FDA呈交有关资料,包括该药物的副作用情况和质量管理记录。对于有些药物FDA还会要求做第四期临床试验,以观测其长期副作用情况。
SMO
到批准上市为止,一个完整的新药研发过程就结束了。但是你会疑惑,为什么临床研究上的描述反而少了。其实是那部分展开来太多了,大大小小几十个甚至上百个流程。从结果论上你也能发现它为什么重要,这里已经是到了人体实验的步骤了,特别是药代动力学、药效动力学和急性毒性实验,这几个最直观也是最后批准生产的一个最低底线。直观的话说,起码你要能完整的在人体产生作用并代谢出去,而且产生的毒性在合理范围。
但近年来为什么医药事故频发呢?直接原因是药物问题,往下一步推导是药物实验漏洞,再往下就是实验流程不严谨和出现漏洞。在中国你可能就赔款了事,在美国那就是捅破天了,除了巨额赔偿、还有罚款以及药物禁入,数十亿美元就化为乌有了。
所以现在中国对标美国,在CRO下面还有一个SMO(临床管理组织),作为实验管理的质量保证。SMO会有很多签了合作协议的PI,就会把任务分劈下去,每个PI按照规范找患者、签协议、试药并监测、填写原始病历报告表等...同时SMO会培训/派驻Study Coordinator到临床点进行协助减少PI的工作量(完成记录、数据输入等辅助工作,确保PI工作量和平时正常做临床差不多,要不PI还要自己做底稿录数据,太繁琐了),最后把数据汇总统计。这里我们就能看到,现在是连实验流程都得标准细致严谨,比如400个高血压病人的标准实验,过程中病人的服药时间、睡眠状况、服药间隔有那么10来个人发生了变化,那么对整个过程的影响都可能是致命的。
现在的临床实验就不再像过去纯以结果导向,过程重视、安全性和药效也重视,要把风险在每一个研发流程都确保消除和可控。但凡任意一个环节出现了错误,都面临推倒重来的风险,不可谓说不细致严谨到了一个高度。
总结:新药研发难不仅仅在于它的时间长、成本高、技术难,更多的还是一个面临推倒重来或者直接失败的风险。复盘过往的全球十大药企,你就能明白它们对于新药研发为什么是又爱又怕了。最后顺带提一提之前说的泰格医药,看好它的原因在于它的核心竞争力和业绩确定性,以及它是目前唯一一家能独立完成临床CRO全流程的企业。