从前年底开始MNC疯狂扫货adc(预算本身也会集中每年年底花),就开始掀起adc浪潮。之前一直没有评论过,一直在学习,随便讲讲一些学习理解感悟,不准确的纠正。
adc就是是抗体+连接子+毒素,分别解决三个药物关键问题,哪里释放、什么时候释放、释放后怎么杀伤。 精准的意义在于能够扩大毒素的治疗窗也就是用量,甚至能够用原先不能直接用的毒素。从这个角度看,抗体偶连这更像一个理念,就是如何把有效的药物送到特定的地方,解决原来小分子药物或者化疗药物一个精准的问题,在这个理念的框架下有很多类似结构的药物思路,目前最广泛成熟的adc 是特定的靶向单抗+小分子,当然小分子也可以换成激动剂、小核酸、核药之类,靶向也可以是双抗、病毒、片段,玩法多种多样,可探索空间还有很大很大。
ADC 替代化疗是大趋势,目前肿瘤领域由于神药8201的出现,大家都冲进去想占份额,尤其是对于中国药企来讲,adc逻辑清晰更像是排列组合和不断的分子筛选过程,不涉及到靶点生物学研究,更具有行业成本优势,造成了火热的现状。
但本质来说,当偶联的是小分子药物的时候,adc本质还是化疗,核心是毒素,靶点只是途径,所以adc不光是要同靶点之间的比,也要和跨靶点同适应症比,头对头更优才有意义。因为前线用过adc后线再用另外一款同毒素的成功率并不大,如果两个adc靶点不同,但是用的毒素原理近似,那么在同一个癌种内互相会不会存在耐药?把adc比作巡航导弹,知道要打击的目标建筑物同时有红色外墙和尖顶这2个识别特征,分别以这2个特征制导,都是能命中目标的,弹药不换的话两者没有实质区别。尤其是现在追求旁观者效应杀伤,到达肿瘤细胞附近后毒素可以进入周围细胞发挥作用,重复靶向差别不大了。
先发优势占赛道也变得尤为重要,前面adc越神药,意味着后续每个adc研发难度越高,不仅mnc买了很多后面有退货是必然,我觉得从大面上看adc的平均研发成本甚至远高于前几代的modality。
那药企现在研发推进adc的思路是什么样呢,从未解决的癌症倒推应该找什么靶点,或者正向的从已知靶点去看有什么适应症可以做、怎么做也是可以的,正推从人家已知的靶点做me-better的平台相对来说是更简单也经济高效的选择方式。
靶点怎么选,适合的适应症是什么?
靶点要满足几个条件:在想要的癌种里高表达,不想要的癌种内低表达,靶点没有太高的靶向毒性,有比较好的内吞效果(这点目前倒也不是必然了,比如宜联的平台专门就是靶向微环境的,就可以做内吞效率一般的靶点比如MET)。目前很成熟经过验证的靶点像HER2(尤其扎堆,但是尤其的没希望![]() )、TROP2、CLDN,B7H3(这个还未成药,但是已经是大热门的adc临床管线),c-MET、nectin-4。
)、TROP2、CLDN,B7H3(这个还未成药,但是已经是大热门的adc临床管线),c-MET、nectin-4。
不是每一个靶点都能成药的,比如一个经典反例EGFR,是一个非常成熟的靶点,但是这个靶点原来做adc一直不成功,直到百利有一线希望。我觉得比较大原因是,EGFR这个相关药物主要分为两类,一是针对下游信号通路中关键蛋白发生突变,导致不依赖EGFR的持续激活,这一类情况主要由TKI药物,比如最经典的奥西替尼来解决,ADC无法直接应对这类驱动基因突变,能通过表面表达的EGFR蛋白来交叉解决耐药问题;二是当细胞表面EGFR过表达,容易激活下游通路,相应的有西妥昔单抗,但是EGFR在普通组织表达太广,西妥昔单抗的策略是只结合在肿瘤细胞表达的EFGRvIII突变型。艾伯维最早的EGFR ADC就是和EGFR结合,但是选择了低活性的抗体,牺牲了毒性导致最后临床失败。艾万妥双抗其中EGFR部分选择亲和力更低,提升C-MET部分活性以C-MET为主靶向(不过强生并不是一开始就想这么做的,这个双抗的成药之路极其艰难复杂,MET过表达在基线中很少,普遍是用药的旁路激活导致)。
确认目标靶点后,再看靶点在哪些肿瘤细胞上存在表达,以TROP2为例,TROP2在正常组织中有表达,在多种上皮来源肿瘤中则是显著高表达的,如乳腺癌(>90%)、尿路上皮癌(<83%)、胃癌(56%)、结肠直肠癌(68.4%)、某些肺癌、胰腺癌、前列腺癌、宫颈癌、头颈癌和卵巢癌等。具体看数据,来源gepia:
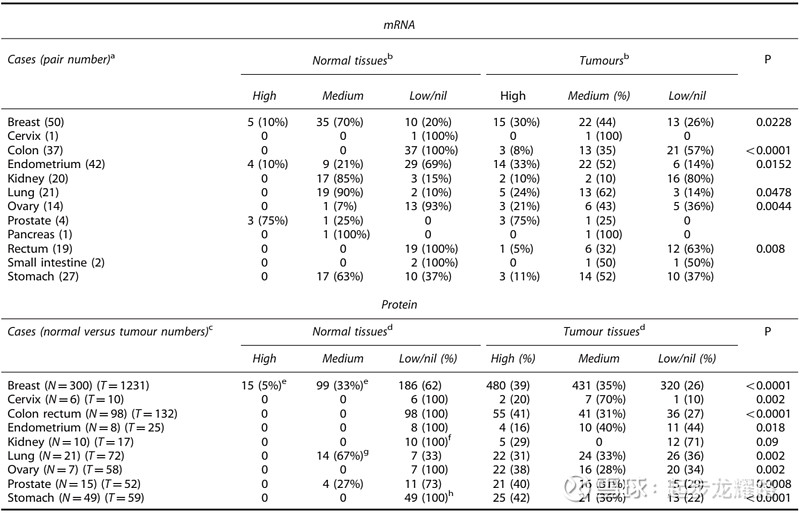
看上去肿瘤细胞和正常细胞表达差异显著的乳腺癌、胃癌、结直肠、卵巢癌都能做。
光确认正常组织表达差异之外,事实上还要看肿瘤细胞上的绝对表达量,一般adc要实现有效的活性,细胞表面抗原的要求为至少需要10^4的绝对表达量,trop2的绝对表达量并不高(HER2在乳腺癌高表达能达到10^6量级,即使是低表达也有10^5,所以所谓ultra-low也是相对的)。网上只找到部分数据:
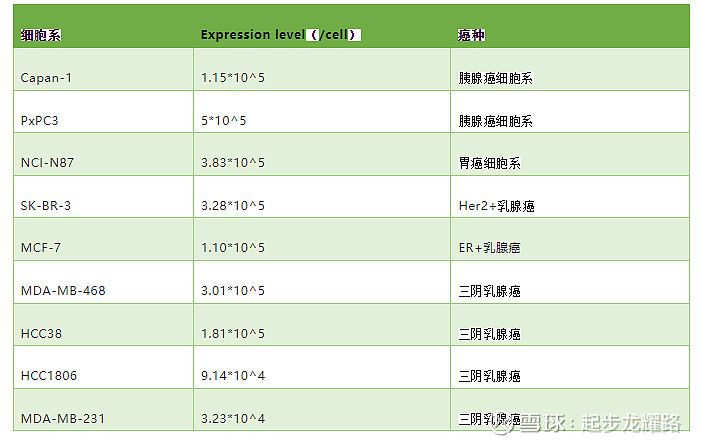
药企内部对绝对表达量肯定会测定,再次筛选能够成药的癌种后,下一步要再看这个适应症原来有哪些用药,以及竞品有哪些adc在研,这直接决定了应该匹配什么样的毒素。
就以最经典的HER2+乳腺癌来说,第一款ADC T-DM1是T(曲妥珠)+DM1(一种微管抑制剂)构成,上市批准是基于III期EMILIA的数据结果,EMILIA入组的是曲妥珠+紫衫类药物(目前标准一线疗法,也可以联用蒽环类阿霉素)治疗后患者,紫衫类药物也是一种微管蛋白抑制剂。即使DM1抗肿瘤活性比紫杉醇强25-500倍,比阿霉素强100-4000倍,可T-DM1的框架没有脱离一线标准曲妥珠+紫衫的思路,临床虽然比拉帕替尼(HER2)联合卡培他滨(DNA毒药物)有显著临床获益,但是OS的HR仅为0.75。所以当T-DXd讲载药换成拓扑异构酶Ⅰ抑制剂DXd(抑制DNA修复解螺旋,抗肿瘤活性是伊立替康活性代谢物SN-38的10倍)的时候,DXd由于不仅具备了旁观者杀伤效应,同时跳出了原先药物机制,头对头T-DM1的临床中PFS的HR直接变成了0.33,提升了惊人的20个月。
说到这里,已经发现有无数个问题需提前审视,1、搭载毒素前线是否有类似药物在用?本身耐药机制?(现在主要就是DNA类毒素和微管抑制剂,一线成熟的化药也是相似逻辑,新毒素还不成熟,所以能理解RDC某种意义作为新毒素就颇受重视。何况ADC本身还有极其复杂耐药机制,参考网页链接 ,MMAE/DM1都是P-gp底物);2、对特定的肿瘤细胞活性,毒素ic50有多高?最合适的毒素小分子是什么?(喜树碱DXd不是对所有细胞系都一定适用,前线伊立替康会不会导致DXd耐药也未可知。第一三共曾经做过依喜替康Exatecan与吉西他滨联用对比吉西他滨单药,联用效果基本等于单药但毒性远大于单药);3、比如对于HER2+乳腺癌适应症,TROP2靶点还有没有意义?(显然没有,所以TROP2才会去做三阴性乳腺癌,甚至恒瑞开了个卵巢癌);4、都说adc可以+pd1,毒素都有刺激细胞死亡的抗原呈递的作用吗?;5、如果自己产品开发的慢了,竞品adc已经上市,临床三期头对头成本吃得消吗?6、靶点表达特异性和毒素毒性、DAR值的平衡关系如何拿捏?等等等等。
我甚至还没提到linker,已经是数不完的组合和坑了。更别提还有无数种其他偶联组合。。
所以启明创投有个观点,对于投资人来讲,靶点和平台不能2个同时都是创新,最好还有安全性数据验证,就比较保险。基于数据驱动的投资比基于原理推测更安全,adc就像大乱炖,所有要素牵一发而动全身环环相扣,不上人体也不能完美预测结果,不要太高预期,而且研发一定是越快越好,这不是一个小biotech能玩的起游戏。